La diabetes tipo 2 contribuye a un elevado riesgo cardiovascular. Un componente importante de dicho riesgo puede atribuirse a la dislipidemia diabética, una agrupación de anomalías de los lípidos y las lipoproteínas plasmáticos que están interrelacionadas metabólicamente. Sus principales características son una elevación de los triglicéridos, una reducción de las lipoproteínas de alta densidad (HDL) y un aumento de las lipoproteínas de baja densidad (LDL) pequeñas y densas, a lo que se denomina la “tríada lipídica aterogénica”. La dislipidemia diabética se asocia a resistencia a la insulina, obesidad visceral y contenido de grasa hepática. La resistencia a la insulina está relacionada con un flujo excesivo de sustratos (es decir, ácidos grasos libres, FFA) para la formación de lipoproteínas de muy baja densidad (VLDL) en el hígado, así como una regulación positiva de los mecanismos que generan partículas VLDL1 grandes en exceso. La identificación de que la elevación de las partículas de VLDL1 grandes inicia una secuencia de episodios que conduce a la formación de especies moleculares de HDL y LDL pequeñas y densas ha hecho que se centrara la atención en las partículas VLDL como posibles responsables de la dislipidemia diabética. Otros componentes del perfil lipídico aterogénico son la apoB elevada, la lipemia posprandial prolongada y la acumulación de partículas residuals en la circulación. Diferentes ensayos prospectivos han demostrado que las estatinas son eficaces en el tratamiento de la dislipidemia diabética, pero el riesgo cardiovascular residual continúa siendo elevado. Es de esperar que en futuros ensayos con nuevos fármacos hipolipemiantes y antidiabéticos permitan realizar un tratamiento más dirigidode los trastornos lipídicos y la lipemia posprandial en los individuos con diabetes tipo 2.
Palabras clave
La diabetes mellitus contribuye de manera importante a la carga total de enfermedades, y se estima que su prevalencia mundial es de 170 millones de casos, cifra ésta que se prevé que aumente a más del doble al llegar al año 20301. La mayor parte de los casos corresponden a la diabetes tipo 2, que se asocia a un exceso de costes de asistencia sanitaria, debidos en gran parte a las complicaciones de enfermedad cardiovascular (ECV) aterosclerótica. En los individuos diabéticos, la enfermedad coronaria (EC) es la causa de hasta un 70%-80% del total de muertes2,3.
Los principales factores de riesgo cardiovascular tradicionales, como hipertensión, tabaquismo y estado procoagulante, explican tan sólo en parte el aumento de riesgo de ECV en la diabetes tipo 2. La hiperglucemia crónica ejerce un efecto nocivo sobre la pared vascular y, a través de la glicación de las apolipoproteínas, interfiere en las vías normales del metabolismo de las lipoproteínas4. Además, en la actualidad hay una evidencia abundante que indica la gran importancia de la dislipidemia como causa de ECV en estos pacientes5–7. Esta breve revisión pretende resaltar algunos aspectos del metabolismo de los lípidos y las lipoproteínas en la diabetes tipo 2, con especial referencia a los cambios que hacen que el perfil lipídico de estos pacientes sea intensamente aterogénico.
CARACTERÍSTICAS GENERALES DEL METABOLISMO LIPÍDICO EN LA DIABETES MELLITUS TIPO 2La dislipidemia diabética es una agrupación compleja de anomalías lipídicas y lipoproteicas potencialmente aterogénicas, que comporta cambios tanto cuantitativos como cualitativos. Sus principales componentes son los siguientes: aumento de los triglicéridos plasmáticos, concentración baja de colesterol de lipoproteínas de alta densidad (C-HDL), predominio de lipoproteínas de baja densidad (LDL) pequeñas y densas, y lipemia posprandial excesiva. Tal como se ha demostrado recientemente, las anomalías del metabolismo lipídico no son cambios aislados sino que están estrechamente relacionadas entre sí7.
El metabolismo lipídico en la diabetes tipo 2 es modulado por una serie de factores entre los que se encuentran el grado de control de la glucemia y la presencia de resistencia a la insulina como elementos más destacados. La resistencia a la insulina está en la base de los mecanismos fisiopatológicos de la dislipidemia diabética, y está estrechamente relacionada con la hipertrigliceridemia y la lipemia posprandial. Puede observarse una hipertrigliceridemia en un 30-60% de los pacientes con diabetes tipo 2, y esta alteración se da con frecuencia en el estado prediabético, precediendo en el tiempo a la aparición de la hiperglucemia crónica8,9. Una importante consecuencia de la resistencia a la insulina respecto al metabolismo lipídico es la pérdida del efecto supresor de la insulina sobre la movilización de la grasa del tejido adiposo. Como consecuencia de ello se produce un aumento del flujo de ácidos grasos libres (FFA) debido a la reducción de la supresión de la lipólisis10. La falta de supresión de los FFA en el período posprandial, como consecuencia de la disminución de actividad de la lipoproteinlipasa (LPL), y el aumento de los FFA plasmáticos a causa de un aumento de la lipólisis en los adipocitos son mecanismos clave que subyacen en el aumento de secreción hepática de triglicéridos (TG) de lipoproteínas de muy baja densidad (TG-VLDL)11.
Los resultados de algunos estudios recientes12,13 han ayudado a aclarar los pasos de la regulación del metabolismo de las VLDL en el hígado en presencia de una diabetes tipo 2 (para una revisión exhaustiva, véanse las referencias 7 y 13). En los individuos normales, la insulina inhibe el ensamblaje y la secreción de las partículas VLDL a través de un aumento de la degradación de la apolipoproteína B (apoB) y una disminución de la expresión de la proteína de transferencia microsómica (MTP) en los hepatocitos14,15. Como consecuencia de ello, la insulina inhibe la secreción hepática de TG-VLDL y de apoB-100.
En la diabetes tipo 2 y otros estados de resistencia a la insulina, se produce en el hígado un aumento de expresión de MTP junto con un aumento de la biodisponibilidad de lípidos (es decir, del flujo de FFA), y ello conduce a una sobreproducción de TG-VLDL y de VLDL-apoB16. En este contexto, la sobreproducción de VLDL hepática corresponde principalmente a partículas VLDL1 grandes y flotantes, que son una característica predominante de la dislipidemia diabética. De hecho, la mayor parte del aumento de lipoproteínas ricas en triglicéridos (TRL) que se observa en la dislipidemia diabética se debe a partículas VLDL112. Además, el hecho de que la insulina no logre inhibir la liberación de VLDL1 en la fase posprandial puede saturar las vías lipolíticas y contribuye a producir la lipemia posprandial.
ALTO RIESGO DE DISFUNCIÓN LIPOPROTEICA EN LA DIABETES MELLITUS TIPO 2Además de lo que se ha descrito, recientemente se han identificado otros factores que pueden contribuir a producir las manifestaciones específicas de la dislipidemia diabética17.
Las vías catabólicas, o lipolíticas de las TRL dependen de 2 enzimas, la lipoproteinlipasa (LPL) y la lipasa hepática (HL), que desempeñan un papel importante en la regulación de las concentraciones plasmáticas de TRL. La insulina ejerce un efecto significativo de estimulación de la actividad de la LPL18. En muchos estudios se ha descrito que la actividad de LPL es baja en los individuos con diabetes tipo 2, mientras que la de la HL está aumentada19. Estos cambios, atribuidos a la resistencia a la insulina, contribuyen a producir un deterioro de la eliminación posprandial de TG y un aumento de la lipemia postprandial que es característica de la dislipidemia diabética.
Apolipoproteína CIII y apolipoproteína ELa apolipoproteína CIII (apoCIII) es la apolipoproteína más abundante en las partículas VLDL y presenta una estrecha correlación con las concentraciones de triglicéridos totales y de VLDL en suero20. La apoCIII modula el metabolismo de las TRL en 2 pasos posteriores21. En primer lugar, la apoCIII es un inhibidor de la LPL, y en segundo lugar interfiere en la fijación al receptor mediada por la apolipoproteína E, con lo que retrasa la eliminación de las partículas residuales de VLDL. Una concentración elevada de apoCIII en las lipoproteínas apoB es un componente frecuente de la dislipidemia aterogénica, y podría ayudar a explicar el riesgo de EC asociado a los triglicéridos elevados. Se ha especulado con la posibilidad de que la apoCIII pueda desempeñar un papel especial en cuanto al aumento del riesgo cardiovascular en los pacientes con diabetes, puesto que la resistencia a la insulina o el déficit de ésta en los individuos diabéticos atenúan la regulación negativa de la expresión de apoCIII en el hígado. Además, el polimorfismo del elemento de respuesta a la insulina del promotor del gen de apoCIII influye en la correlación entre insulina y TRL22. Sin embargo, hasta el momento, son pocos los estudios realizados sobre las concentraciones de apoCIII en una población diabética, y los datos existentes no son concluyentes23. Alaupovic et al24 observaron que la apoCIII estaba aumentada en las lipoproteínas apoB de los pacientes con diabetes tipo 2. Sin embargo, un análisis post-hoc más reciente del ensayo CARE25 ha demostrado que los individuos diabéticos no presentaban unas concentraciones de apoCIII superiores a las de los no diabéticos con unos valores de triglicéridos en ayunas similares. Así pues, según este análisis, la diabetes no parece asociarse de por sí a unas concentraciones elevadas de partículas lipoproteicas ricas en triglicéridos con contenido de apoCIII. En la misma línea, un estudio reciente de Hiukka et al26 ha descrito un déficit relativo de apoCIII en todas las especies moleculares de lipoproteínas ricas en triglicéridos en los individuos con diabetes tipo 2, lo cual aporta una evidencia indicativa de que, a pesar del aumento de la concentración plasmática de apoCIII en estos individuos, la concentración de apoCIII no aumenta en paralelo con los lípidos contenidos en la partícula. Dado que la apoCIII desempeña un papel crucial en la eliminación hepática de los residuos de VLDL, esta falta relativa de apoCIII podría dar lugar a un deterioro en la eliminación de dichas partículas y a un tiempo de permanencia prolongado en la circulación, agravando con ello el riesgo de ECV.
Los polimorfismos de apolipoproteína E (apoE) pueden influir en las relaciones claramente establecidas de la hipertrigliceridemia con la hiperinsulinemia en los trastornos de resistencia a la insulina/hiperinsulinemia. Se ha demostrado que, a diferencia de las portadoras de apoE2 y apoE3, que muestran unas concentraciones plasmáticas de triglicéridos más altas con la hiperinsulinemia en ayunas, las mujeres premenopáusicas sanas con la isoforma apoE4 tienen unas concentraciones plasmáticas de triglicéridos similares a concentraciones altas y bajas de insulina en ayunas27.
FRACCIONES DEL COLESTEROLLos valores bajos de colesterol HDL, que son un factor de riesgo de ECV bien conocido, constituyen otra característica destacada de la dislipidemia diabética y se acompañan de una desviación hacia un tamaño de las partículas más pequeño7. El aumento de los triglicéridos plasmáticos comporta un intercambio de lípidos centrales entre las partículas TRL y las HDL. Se produce un aumento de la transferencia de colesterol esterificado a las TRL, facilitado por la proteína de transferencia de éster de colesterol (CETP), y de la transferencia de triglicéridos a las partículas HDL, con lo que se produce un enriquecimiento de triglicéridos en estas últimas. Los triglicéridos de las HDL son un sustrato adecuado para la lipasa hepática, y la hidrólisis produce unas partículas HDL más pequeñas y apoAI libre que son excretadas por los riñones28. El catabolismo de las HDL pequeñas es más rápido que el de las HDL normales, y ello da lugar a una reducción de la cantidad de partículas HDL circulantes. Así pues, en la patogenia de la reducción de las HDL desempeñan un papel básico tanto el enriquecimiento de las HDL en triglicéridos como el aumento de la actividad de lipasa hepática, que da lugar a un aumento de la rapidez de eliminación de las partículas HDL y una reducción de éstas. El efecto protector de las HDL se atribuye principalmente a su papel en la inversión del transporte del colesterol, pero es posible que intervengan también en ello otras propiedades de las HDL (antiinflamatorias, antioxidantes, antitrombóticas, etc.). Cabe concluir que las alteraciones de las HDL observadas en la dislipidemia diabética (menor tamaño y concentraciones más bajas) contribuyen a producir el aumento de riesgo cardiovascular en los diabéticos tipo 2.
Las partículas de LDL pequeñas y densas (fenotipo B) son una característica prominente de la dislipidemia diabética. El número de estas partículas aterogénicas está aumentado pero la concentración de C-LDL continúa siendo normal o incluso inferior a la normal. Así pues, en este contexto, la concentración absoluta de CLDL puede ser un dato que lleve a engaño puesto que no refleja la presencia de un aumento en el número de partículas aterogénicas29.
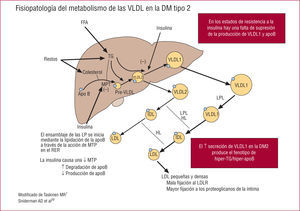
Se ha confirmado repetidas veces que la concentración de triglicéridos plasmáticos es el factor determinante más importante del tamaño de las LDL30. Por otra parte, el tamaño de las LDL disminuye de manera escalonada con la progresión de la tolerancia normal a la glucosa a la intolerancia a la glucosa y a la diabetes manifiesta, y el descenso es mayor en las mujeres que en los varones31. Los datos del grupo de Taskinen12 indican que los triglicéridos de VLDL1 son el principal predictor del tamaño de las LDL en los individuos con diabetes tipo 2, y los datos cinéticos indican que las VLDL1 son los precursores de las partículas de LDL pequeñas y densas (fig. 1). De hecho, el largo tiempo de permanencia de las VLDL1 en el plasma, debido a la reducción de la lipólisis, es un requisito previo para la formación de LDL pequeñas y densas, puesto que favorece el exceso de intercambio lipídico de TG y ésteres de colesterol entre TRL y LDL. Cuando las LDL han sufrido una depleción de ésteres de colesterol y un enriquecimiento en TG, el aumento de la acción de la lipasa hepática da lugar a la formación de la subclase de partículas pequeñas y densas. Dado que cada partícula de LDL contiene una molécula de apoB100, el número de LDL pequeñas y densas está aumentado y, de igual modo, la concentración de apoB100 aumenta en relación directa con ello. En consecuencia, las concentraciones de apoB son un marcador del número de partículas aterogénicas y la hipertrigliceridemia con hiper-apoB100 es una característica bien conocida de la dislipidemia diabética y otros estados de resistencia insulínica29. En esta misma línea, la evidencia basada en estudios básicos, epidemiológicos y de ensayos clínicos indica que la determinación de la apoB en plasma es un indicador fiable del riesgo cardiovascular y debe incluirse en todas las directrices32.
 y se produce a través de la acción de la proteína de transferencia de triglicéridos microsómica (MTP). En los estados fisiológicos, la insulina inhibe la acción de la MTP y reduce la síntesis de VLDL. Esta acción falla en los estados de resistencia a la insulina. FFA: ácidos grasos libres; HL: lipasa hepática; IDL: lipoproteínas de densidad intermedia; LDL: lipoproteínas de baja densidad; LDLR: receptor de las lipoproteínas de baja densidad; LPL: lipoproteinlipasa.")
Papel de la insulina en la regulación de la síntesis de VLDL en el hígado. El ensamblaje de las partículas VLDL empieza en el retículo endoplásmico rugoso (RER) y se produce a través de la acción de la proteína de transferencia de triglicéridos microsómica (MTP). En los estados fisiológicos, la insulina inhibe la acción de la MTP y reduce la síntesis de VLDL. Esta acción falla en los estados de resistencia a la insulina. FFA: ácidos grasos libres; HL: lipasa hepática; IDL: lipoproteínas de densidad intermedia; LDL: lipoproteínas de baja densidad; LDLR: receptor de las lipoproteínas de baja densidad; LPL: lipoproteinlipasa.
Las partículas LDL modificadas y pequeñas y densas son más aterógenas por una serie de razones33. La mala unión al receptor de LDL favorece un tiempo de permanencia prolongado y esto, junto con el menor tamaño de la partícula, facilita la penetración en la íntima arterial34. Además, las LDL pequeñas y densas tienen una mayor afinidad de fijación que las partículas de mayor tamaño a los proteoglicanos de la íntima de la pared arterial, y son un objetivo fácil para las modificaciones oxidativas35.
En la dislipidemia diabética, la asociación entre acción de la insulina y metabolismo lipídico podría verse influida también por factores que afectan al metabolismo de la glucosa y los lípidos, como la obesidad, la distribución abdominal de la grasa, la actividad física, la dieta, el hábito tabáquico, la edad, el sexo, etc. La cantidad de grasa visceral es un factor importante para la modulación del grado de sensibilidad a la insulina y el perfil lipídico, y puede explicar las importantes diferencias metabólicas que hay entre los varones y las mujeres36. La obesidad se da con gran frecuencia en los diabéticos tipo 2 y su influencia en la resistencia a la insulina y la dislipidemia diabética está bien establecida. En diferentes estudios se ha demostrado que la sensibilidad a la insulina periférica, expresada mediante el índice Si, está reducida de manera significativa en los individuos con obesidad visceral que presentan unos FFA plasmáticos elevado37.
En resumen, la dislipidemia diabética es una agrupación de anomalías lipídicas que están estrechamente relacionadas entre sí desde el punto de vista metabólico. Se ha revisado brevemente la hipertrigliceridemia, debida en su mayor parte a una producción excesiva de partículas VLDL1 grandes y flotantes, el C-HDL bajo, el predominio de la subclase de partículas LDL pequeñas y densas y la lipemia posprandial prolongada, y se ha resaltado el importante papel que desempeña la resistencia a la insulina. Los componentes aterogénicos más potentes de la dislipidemia diabética son las LDL pequeñas y densas, la elevación de las partículas residuales de TRL y las HDL bajas. La coexistencia de estos tres factores agrava intensamente la acumulación de lípidos en la pared arterial y la formación de placas ateroscleróticas.
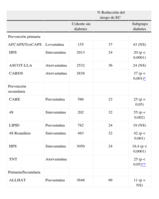
EFECTOS DE LOS TRATAMIENTOS MODIFICADORES DE LOS LÍPIDOS SOBRE LAS ANOMALÍAS LIPÍDICAS EN LA DIABETES TIPO 2Además de los cambios de estilo de vida y la optimización del control diabético con fármacos orales y/o insulina, los individuos con dislipidemia diabética necesitarán un tratamiento con fármacos hipolipemiantes destinado a reducir su gran carga de ECV. En la tabla 1 se resumen los resultados de algunos ensayos prospectivos de prevención primaria y secundaria con estatinas en los que se incluyó a pacientes diabéticos. Para una revisión exhaustiva de estos ensayos, véanse las referencias38,39.
Análisis de subgrupos en ensayos de prevención de la EC con estatinas en pacientes diabéticos
| % Reducción del riesgo de EC | ||||
| Cohorte sin diabetes | Subgrupo diabetes | |||
| Prevención primaria | ||||
| AFCAPS/TexCAPS | Lovastatina | 155 | 37 | 43 (NS) |
| HPS | Simvastatina | 2913 | 24 | 20 (p<0,0001) |
| ASCOT-LLA | Atorvastatina | 2532 | 36 | 24 (NS) |
| CARDS | Atorvastatina | 2838 | 37 (p=0,001)* | |
| Prevención secundaria | ||||
| CARE | Pravastatina | 586 | 23 | 25 (p=0,05) |
| 4S | Simvastatina | 202 | 32 | 55 (p=0,002) |
| LIPID | Pravastatina | 782 | 24 | 19 (NS) |
| 4S Reanálisis | Simvastatina | 483 | 32 | 42 (p=0,001) |
| HPS | Simvastatina | 3050 | 24 | 18,4 (p<0,0001) |
| TNT | Atorvastatina | 25 (p<0,05)** | ||
| Primaria/Secundaria | ||||
| ALLHAT | Pravastatina | 3648 | 99 | 11 (p=NS) |
Adaptado y modificado de Carmena R38.
Tal como se indica en la tabla 1, se ha demostrado que el tratamiento con estatinas reduce significativamente el riesgo de ECV y se considera un tratamiento hipolipemiante de primera línea en los pacientes con diabetes17,38,39. La reducción del riesgo de ECV se asocia a una reducción de las concentraciones plasmáticas de C-LDL de un 25-35%. En el subgrupo de pacientes diabéticos del ensayo Treating to New Targets (TNT)40, el tratamiento hipolipemiante intensivo con atorvastatina 80mg/día aportó un beneficio clínico significativo que iba más allá del proporcionado por atorvastatina 10mg/día en pacientes con una EC estable. Una reducción de la concentración media de C-LDL de 2,0mmol/l con atorvastatina 80mg/día se asoció a una reducción del 25% en el riesgo relativo de episodios cardiovasculares en comparación con lo observado con una reducción del C-LDL medio de 2,6mmol/l con atorvastatina 10mg/día (p<0,05). Así pues, la reducción del C-LDL hasta valores significativamente más bajos que los actualmente recomendados produjo una reducción sustancial de los episodios CV primarios en este grupo de pacientes diabéticos. Se observaron también diferencias significativas entre los grupos, favorables a atorvastatina 80mg, en cuanto al tiempo transcurrido hasta los episodios cerebrovasculares, con una reducción del riesgo relativo de un 31% (p<0,05).
A pesar de los efectos beneficiosos concluyentes aportados por las estatinas, el riesgo cardiovascular residual en los pacientes con diabetes tipo 2 continúa siendo elevado. Parece que podría haber un límite en el grado de beneficio que puede obtenerse con la sola reducción de las concentraciones de C-LDL, y el riesgo residual se ha atribuido, al menos en parte, a la ausencia de elevación del C-HDL. De hecho, los datos de los ensayos clínicos realizados en pacientes con diabetes tipo 2 han demostrado que las estatinas no son capaces de eliminar el riesgo asociado al C-HDL bajo. En el Heart Protection Study41, a pesar de las reducciones uniformes del C-LDL y la apoB, el riesgo de episodios de EC continuó siendo significativamente más alto en los individuos diabéticos con un C-HDL inferior a 0,9mmol/l tratados con simvastatina, en comparación con el de los individuos con un C-HDL normal que recibieron placebo, y se han descrito unos resultados similares en el ensayo CARE42. Así pues, el aumento de las HDL ha pasado a ser un objetivo importante para optimizar la reducción del riesgo en la dislipidemia diabética. Recientemente se ha interrumpido de manera prematura un ensayo prospectivo con torcetrapib, un inhibidor de la proteína de transferencia de ésteres de colesterol (CETP) capaz de elevar de manera significativa el C-HDL43, debido a un aumento inesperado de la mortalidad total. Sin embargo, actualmente se están contemplando otras estrategias para la elevación del C-HDL, como la modulación del receptor nuclear y la infusión de complejos de apolipoproteínas/ fosfolípidos44.
A pesar de los efectos beneficiosos de los fibratos en los individuos con diabetes tipo 2 que se han descrito en algunos ensayos clínicos45, un análisis global de todos los ensayos importantes realizados con fibratos muestra unos resultados poco homogéneos46. En el estudio FIELD47, el mayor ensayo prospectivo realizado hasta el momento sobre los lípidos en varones y mujeres con diabetes tipo 2, con o sin ECV previa, el tratamiento con fenofibrato no produjo unos resultados tan favorables como se esperaba. En el conjunto de la población, el tratamiento con fenofibrato se asoció a una reducción no significativa del 11% en la variable de valoración principal (muerte por EC e infarto de miocardio no mortal). El total de episodios de ECV se redujo en un 19% (p=0,004) en los pacientes sin ECV previa, pero, sorprendentemente, no ocurrió así en los pacientes sin ECV previa, en los que se observó un aumento. No disponemos de una explicación clara para estos resultados, pero se ha considerado la posibilidad de que la activación crónica del PPARα producida por fenofibrato en el músculo cardíaco pudiera tener efectos nocivos17. Además, a diferencia de lo observado en los ensayos con estatinas, el ictus no se redujo de manera significativa en el estudio FIELD. Es interesante señalar que el efecto de elevación del C-HDL producido por fenofibrato se atenuó a lo largo del período de estudio y que la diferencia entre los grupos de fenofibrato y placebo era trivial al finalizar el estudio. No obstante, algunos investigadores creen que los fibratos podrían continuar teniendo un papel selectivo en el tratamiento de los pacientes con hipertrigliceridemia intensa, en especial en combinación con un tratamiento de estatinas46. Es de esperar que la cuestión de si la combinación de una estatina con un fibrato aporta o no un efecto beneficioso adicional que va más allá del obtenido con la estatina sola pueda ser resuelta por el estudio Action to Control Cardiovascular Risk in Diabetes (ACCORD) actualmente en marcha (http://accordtrial.org/public/index.cfm).
Recientemente, varios estudios de corta duración con otros productos, como agonistas de PPARγ 48,49, bloqueantes de receptores CB1 de endocannabinoides50 o sustancias del nuevo grupo de fármacos de incretina51, han descrito una influencia favorable en la dislipidemia diabética y la lipemia postprandial.nque estos resultados son alentadores, serán necesarios nuevos estudios para poder alcanzar conclusiones definitivas. Es preciso explorar también los efectos beneficiosos plenos que se obtienen con la combinación de estos fármacos con estatinas. Así pues, es de esperar que en un futuro próximo pueda disponerse de un tratamiento más dirigido de las anomalías lipídicas en la diabetes tipo 2, con una mayor reducción concomitante del riesgo de EC.
CONCLUSIONESEn resumen, se han revisado las complejidades de las disfunciones de las lipoproteínas en la diabetes tipo 2. Los estudios cinéticos recientes sobre el papel de la insulina en el ensamblaje hepático de las VLDL y el metabolismo periférico de estas partículas han aportado datos importantes y han ampliado nuestro conocimiento de esta forma aterógena de dislipidemia. Se espera que el mejor conocimiento de los factores patogénicos que intervienen en la disfunción de las lipoproteínas observada en estos pacientes se traduzca en la disponibilidad de tratamientos más eficaces y en un beneficio clínico sustancial.
AGRADECIMIENTOSEste trabajo fue financiado por una subvención del Ministerio de Sanidad de España, Instituto de Salud Carlos III (Madrid), Red Diabetes (REDIMET # RD06/0015/0015).
El autor no declara ningún conflicto de intereses.