
Dilated cardiomyopathy is inherited in nearly 50% of cases. More than 90 genes have been associated with this disease, which is one of the main causes of heart transplant and has been associated with an increased risk of sudden cardiac death. Risk stratification in these patients continues to be challenging. The identification of the specific etiology of the disease is very useful for the early detection of mutation carriers. Genetic study often provides prognostic information and can determine the therapeutic approach. Wide phenotypic variability is observed depending on the mutated gene, the type of mutation, and the presence of additional genetic and environmental factors.
Keywords
Dilated cardiomyopathy (DCM) is defined as dilatation and systolic dysfunction of the left ventricle or both ventricles in the absence of abnormal loading conditions or heart disease.1 DCM is one of the main causes of heart transplantation and is associated with an increased risk of sudden cardiac death. The prevalence of DCM is estimated at 1 in 2500 individuals, but recent studies suggest it might be higher.2 DCM is inherited in an estimated 50% of cases, and research in recent years has identified more than 90 genes implicated in the disease (Table 1). Most heritable forms of DCM are autosomal dominant, whereas X-linked, autosomal recessive, and mitochondrial forms are less frequent. The implicated genes encode sarcomere and cytoskeletal components, intercellular junctions, ion channels, and mitochondrial proteins.
Genes associated with dilated cardiomyopathy. Priority genes are clearly associated with disease and are covered by clinical practice guidelines; secondary genes have been sporadically associated with disease; and candidate genes have been identified by systematic review of the medical literature.
| Gene | Protein | Category |
|---|---|---|
| TTN | Titin | Priority |
| LMNA | Lamin A/C | Priority |
| DMD | Dystrophin | Priority |
| MYH7 | Beta-myosin heavy chain | Priority |
| DSP | Desmoplakin | Priority |
| BAG3 | BAG family molecular chaperone regulator 3 | Priority |
| FLNC | Filamin C | Priority |
| ACTC1 | Actin | Priority |
| RBM20 | RNA-binding motif protein 20 | Priority |
| TNNT2 | Troponin T | Priority |
| MYBPC3 | Myosin-binding protein C, cardiac-type | Priority |
| PKP2 | Plakophilin | Priority |
| PLN | Phospholamban | Priority |
| DES | Desmin | Priority |
| TNNI3 | Troponin I | Priority |
| TNNC1 | Troponin C | Priority |
| TPM1 | Tropomyosin | Priority |
| TAZ | Tafazzin | Priority |
| ABCC9 | ATP-binding cassette subfamily C member 9 | Secondary |
| ACTA1 | Alpha actin 1 | Secondary |
| ACTN2 | Alpha actinin 2 | Secondary |
| ALMS1 | Alstrom syndrome protein 1 | Secondary |
| ANKRD1 | Ankyrin repeat domain 1 | Secondary |
| ANO5 | Anoctamin 5 | Secondary |
| CAV3 | Caveolin 3 | Secondary |
| CHRM2 | Cholinergic receptor, muscarinic 2 | Secondary |
| COL7A1 | Collagen type VII, alpha 1 chain | Secondary |
| CRYAB | Alpha-crystallin B chain | Secondary |
| CSRP3 | Cysteine and glycine-rich protein 3 | Secondary |
| DNAJC19 | Mitochondrial import inner membrane translocase subunit TIM14 | Secondary |
| DOLK | Dolichol kinase | Secondary |
| DSC2 | Desmocollin 2 | Secondary |
| DSG2 | Desmoglein 2 | Secondary |
| EMD | Emerin | Secondary |
| EYA4 | Eyes absent homolog 4 | Secondary |
| FHL2 | Four and a half LIM domains protein 2 | Secondary |
| FHOD3 | Formin Homology 2 Domain Containing 3 | Secondary |
| FKRP | Fukutin related protein | Secondary |
| FKTN | Fukutin | Secondary |
| FOXD4 | FOXD4 transcription factor | Secondary |
| GAA | Glucosidase alpha, acid | Secondary |
| GATA4 | GATA4 transcription factor | Secondary |
| GATA6 | GATA6 transcription factor | Secondary |
| GATAD1 | GATAD1 protein | Secondary |
| GLB1 | Beta galactosidase | Secondary |
| HFE | HFE protein (iron transporter) | Secondary |
| JUP | Plakoglobin | Secondary |
| LAMA2 | Laminin, alpha 2 | Secondary |
| LAMA4 | Laminin, alpha 4 | Secondary |
| LAMP2 | Lysosome-associated membrane protein 2 | Secondary |
| LDB3 | LIM domain binding 3 | Secondary |
| MURC | Caveolae associated protein 4 | Secondary |
| MYH6 | Myosin heavy chain 6 | Secondary |
| MYL2 | Myosin regulatory light chain 2 | Secondary |
| MYL3 | Myosin essential light chain 3 | Secondary |
| MYOT | Myotilin | Secondary |
| MYPN | Myopalladin | Secondary |
| NEBL | Nebulette | Secondary |
| NEXN | Nexilin | Secondary |
| PRDM16 | PRDM16 protein | Secondary |
| PSEN1 | Presenilin 1 | Secondary |
| PSEN2 | Presenilin 2 | Secondary |
| RAF1 | RAF proto-oncogene serine/threonine protein kinase | Secondary |
| RYR2 | Ryanodine receptor 2 | Secondary |
| SCN5A | Sodium voltage-gated channel alpha subunit 5 | Secondary |
| SDHA | Succinate dehydrogenase complex flavoprotein subunit A | Secondary |
| SGCD | Sarcoglycan delta | Secondary |
| SLC22A5 | SLC22A5 protein | Secondary |
| SPEG | Striated muscle enriched Ser/Thr protein kinase | Secondary |
| SYNE1 | Nesprin 1 | Secondary |
| SYNE2 | Nesprin 2 | Secondary |
| TBX20 | TBX20 transcription factor | Secondary |
| TCAP | Telethonin | Secondary |
| TMEM43 | Transmembrane protein 43 | Secondary |
| TMPO | Thymopoietin | Secondary |
| TOR1AIP1 | Torsin 1A interacting protein 1 | Secondary |
| TTR | Transthyretin | Secondary |
| TXNRD2 | Thioredoxin reductase 2 | Secondary |
| VCL | Vinculin | Secondary |
| XK | Membrane transport protein XK | Secondary |
| BRAF | Serine/threonine protein kinase B-raf | Candidate |
| DNM1L | Dynamin 1-like protein | Candidate |
| GATA5 | GATA5 transcription factor | Candidate |
| GLA | Galactosidase alpha A | Candidate |
| IDH2 | IDH2 mitocondrial protein | Candidate |
| ILK | Integrin-linked protein kinase | Candidate |
| KCNJ2 | Potassium voltage-gated channel subfamily J member 2 | Candidate |
| KCNJ8 | Potassium voltage-gated channel subfamily J member 8 | Candidate |
| NKX2-5 | NK2 homeobox 5 transcription factor | Candidate |
| OBSCN | Obscurin | Candidate |
| OPA3 | Optic atrophy 3 protein | Candidate |
| PDLIM3 | PDZ and LIM domain protein 3 | Candidate |
| TPN11 | Tyrosine protein phosphatase 11 | Candidate |
| SGCA | Alpha-sarcoglycan | Candidate |
| SGCB | Beta-sarcoglycan | Candidate |
| TNNI3K | TNNI3-interacting serine/threonine kinase | Candidate |
Risk stratification of DCM patients remains challenging. A variety of factors show an association with increased risk of disease progression and sudden cardiac death; however, these factors are rarely clinically useful, and prognosis is usually determined by the patient's functional class and disease severity assessed by imaging techniques.3 The importance of determining genotype has been established for some specific forms of the disease, such as those involving mutations in lamin A/C (LMNA), which are associated with poor prognosis.4,5 Given that the causes of DCM are so varied, it is probably inappropriate to attempt a generalized assessment of poor prognosis risk in these patients; rather, suitable prognostic stratification and choice of therapeutic strategy will require identification of the specific etiology of each form of the disease. Knowledge of the underlying cause of disease also permits early diagnosis of family members carrying the disease-causing mutation, so that they can be closely monitored and receive early treatment, as well as avoiding unnecessary monitoring of noncarriers.6
This review aims to describe the usefulness of genetic testing in the prognostic assessment of DCM patients. Prognosis will depend not only on the identity of the mutated gene, but also on the nature of the specific mutation within a single gene; indeed, for any gene of interest, there may be variants that do not cause disease. Even within the same family, the same identified mutation can produce different phenotypes due to interactions with other genetic and environmental factors and the influence of age and sex, etc. It is therefore essential to assess each identified genetic variant independently and to conduct a systematic genetic and clinical analysis of families affected by DCM. Reports on disease-associated genes often fail to present detailed patient and family clinical data, and therefore for some genes it is difficult to reach a conclusion on the likely importance of the identified mutations.
DCM CAUSED BY MUTATIONS IN SARCOMERE GENESTitinTTN encodes the largest protein expressed in the heart and is the gene most frequently associated with DCM. The titin protein spans from the Z-disc to the M line in the center of the sarcomere and is responsible for maintaining sarcomere structural integrity. Many TTN mutations are truncating (frameshift mutations, nonsense mutations, and mutations generating RNA splice variants), and many of these truncating TTN mutations show an autosomal dominant association with DCM that explains 14% to 25% of cases of this disease.7,8 Most of the identified disease-causing mutations are located in the titin A-band and affect TTN exons included in many of the different titin isoforms.9 Other phentoypes linked to titin truncations are tibial muscular dystrophy and some recessive diseases, such as type-2 limb-girdle muscular dystrophy and early onset skeletal and cardiac myopathy. An early study of TTN truncations in DCM found no relationship between the presence of TTN mutations and the rate of cardiac outcomes; however, analysis by sex revealed that male mutation carriers had adverse events at a younger age.7 A more recent analysis demonstrated a higher prevalence of ventricular arrhythmia in carriers than in noncarriers (64% vs 21%), albeit in a small study population.10 Larger studies have reported a 3-fold higher risk of atrial fibrillation or ventricular tachycardia after adjusting for conventional risk factors. In one study, arrhythmias were detected in 46% of DCM patients with TTN truncations vs 33% of DCM patients not carrying these mutations.11 Another study reported a more positive prognosis for DCM patients with TTN truncations than for their counterparts with mutations in LMNA or with no identified genetic cause; patients with TTN truncations had less severe disease at presentation and responded more favorably to standard therapy.12 Our recent study presented at the 2018 European Society of Cardiology Congress analyzed data from more than 500 index patients and family members with TTN mutations. The study revealed an elevated rate of cardiovascular death in affected individuals older than 30 years; the mortality rate was higher for men than for women, and sudden cardiac death accounted for half of the events.13
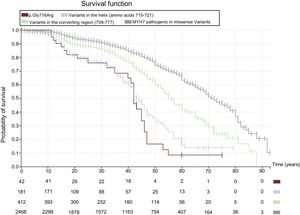
Sarcomere proteinsGenes encoding sarcomere proteins are strongly associated with the development of hypertrophic cardiomyopathy (HCM) through autosomal dominant inheritance.14 However, most of these genes are also implicated in DCM. The most frequently affected genes are MYH7, TNNT2, and TPM1, whereas MYBPC3 gene variants are less common. The presence of these mutations has been linked to the early development of DCM, characterized by early onset heart failure in the absence of previous evidence of hypertrophy or myofibrillar disorganization, thus indicating primary DCM.15 Another study of rare sarcomere gene variants in DCM showed an increase in the rate of events (death and heart transplantation) after the age of 50 years, independently of the ejection fraction; however, carriers and noncarriers showed no difference in overall long-term survival.16 The prognostic impact of MYH7 genetic variants depends on the location of the mutation in the expressed protein. Missense variants in the converter region (amino acids 709-777) have been linked to early disease onset and a high prevalence of events.17 Even within this region, different mutations have markedly distinct effects on disease progression and prognosis. In particular, mutations in the alpha helix spanning amino acids 715 to 722 are associated with an especially poor prognosis, including a high rate of sudden cardiac death among young patients and heart failure leading to death or heart transplant before the age of 50 years (Figure 1). Another functionally important region appears to be the actin-binding domain (amino acids 526-557), with genetic variants identified in 64 carriers from 30 families; most patients with these variants were diagnosed early in life and developed moderate ventricular dysfunction (unpublished data). It is not possible to describe a general prognosis for mutations in TTNT2; however, those that cause DCM tend to be highly penetrant and are associated with a high frequency of adverse events.18 Several missense mutations linked to DCM have been identified in TPM1, clustering in the central flexible region in the C-terminal domain (amino acids 81-258). Many of these mutations have been linked to disease in early childhood, sometimes with events principally triggered by heart failure.19 It should be noted that mutations in MYBPC3 are not generally associated with poor prognosis; however, they can give rise to severe phenotypes in homozygotes or compound heterozygotes.
 Events in carriers of any disease-causing missense mutations (gray) are compared with events associated with the converter region (amino acids 709-711; green), the alpha helix spanning amino acids 715-721 (violet), and the p.Gly716Arg missense variant (brown). The chart reveals significant differences for mutations in different regions, with alpha helix variants such as p.Gly716Arg associated with very poor survival at the age of 50 years. An initial or later diagnosis of dilated cardiomyopathy was more frequent in patients carrying variants linked to a poor prognosis than in those carrying variants linked to a better prognosis.")
Kaplan-Meier curves estimating cardiovascular-death–free survival in carriers of disease-causing missense mutations in MYH7. (Cardiovascular death includes sudden cardiac death, death related to appropriate defibrillator discharge, death due to heart failure or heart transplantation, or death due to any other cardiovascular cause.) Events in carriers of any disease-causing missense mutations (gray) are compared with events associated with the converter region (amino acids 709-711; green), the alpha helix spanning amino acids 715-721 (violet), and the p.Gly716Arg missense variant (brown). The chart reveals significant differences for mutations in different regions, with alpha helix variants such as p.Gly716Arg associated with very poor survival at the age of 50 years. An initial or later diagnosis of dilated cardiomyopathy was more frequent in patients carrying variants linked to a poor prognosis than in those carrying variants linked to a better prognosis.
Actin, encoded by the ACTC1 gene, is the principal component of the thin filaments in the sarcomere. Of the few mutations reported for this gene, some are linked to an overlapping phenotype of noncompaction HCM and autosomal dominant DCM. Some of the described ACTC1 genetic variants are associated with septal defects, and progression to heart failure in these patients is characterized by diastolic dysfunction and a restrictive cardiomyopathy phenotype.20
DCM CAUSED BY MUTATIONS IN CYTOSKELETAL PROTEINSLaminThe LMNA gene encodes 2 proteins, lamins A and C, which are components of the inner nuclear membrane. Mutations in this gene are associated with a group of autosomal dominant inherited diseases known collectively as laminopathies. Laminopathies involving lamin A/C include DCM, Emery-Dreifuss muscular dystrophy, familial partial lipodystrophy, limb-girdle muscular dystrophy, Charcot–Marie–Tooth disease, and progeria. Cardiac involvement is characterized by a high risk of sudden cardiac death and high rates of conduction disorders and ventricular arrhythmias, which generally precede the onset of ventricular dysfunction.21LMNA genetic variants are also frequently associated with supraventricular arrhythmias (atrial fibrillation). Penetrance is very high, with almost 100% of mutation carriers developing the disease by the age of 60 years. A low threshold is recommended for placing an implantable cardioverter-defibrillator (ICD) in these patients, especially those requiring pacing.22,23 The most frequently described genetic variants are missense mutations located in the central domain of the protein; however, there have also been reports of truncation variants. Carriers of the disease-causing mutations have a poor prognosis, but the mutations differ in their effects, and some rare variants do not cause disease. A European cohort study identified 4 factors that independently increase the risk of arrhythmias in mutation carriers: the presence of nonsustained ventricular tachycardia, an ejection fraction <45%, male sex, and nonmissense mutations (nonsense, frameshift, and splicing mutations).22 Nevertheless, in an analysis of data from more than 1000 LMNA mutation carriers and their family members, our group found no major differences in adverse events between men and women (unpublished data). Moreover, preliminary results from a Spanish multicenter study showed no prognostic differences between truncating and missense mutations in LMNA.24 These data indicate the need to revise the criteria currently used to indicate ICD placement in these patients, especially given that sudden cardiac death is not infrequent in patients with an ejection fraction> 45%.25
DesminThe cytoplasmic protein desmin is the major component of intermediate filaments and is encoded by the DES gene. Mutations in DES have been linked to DCM, conduction disorders, and progressive muscle weakness and are highly penetrant. Autosomal recessive and autosomal dominant mutations have been identified, and most of the disease-causing variants arise from missense mutations. The prevalence of desminopathies is approximately 1 in 10 000.26 In the heart, conduction disorders tend to precede myocardial alterations, and the cardiomyopathy is frequently restrictive.27 Up to 50% of carriers develop cardiomyopathy, and approximately 60% have conduction disorders and ventricular arrhythmias. The absence of cardiomyopathy does not equate to absence of disease. Although atrioventricular block is a characteristic feature of desminopathy, fatal arrhythmia has been reported, including in patients fitted with a pacemaker.28
DystrophinDystrophin is a large cytoskeletal protein located on the inner surface of the sarcolemma and is encoded by the DMD gene. Mutations in DMD are associated with 3 phenotypes: Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and X-linked DCM. These phenotypes differ in the severity of muscle involvement. DMD-associated DCM can affect male and female carriers of disease-causing mutations, with diagnosis between the ages of 20 and 40 years for men and a little later for women. Progression to severe disease tends to be rapid in men, whereas it can take several years in women. Cardiac deterioration is detected in 60% to 75% of patients and is characterized by diffuse ventricular degeneration and fibrosis, especially in the inferolateral regions and conduction tissue. The disease frequently features atrial and ventricular arrhythmias. In more than 2 out of every 3 patients, the mutation involves the deletion of 1 or more exons, most often in a specific region between exons 45 and 53. A small proportion of affected individuals (5%-15%) have partial DMD gene duplications. Missense mutations are less frequent and generally affect heart-specific regions of the protein; more than half of the DCM-related missense mutations in DMD are located in the actin-binding domain.29,30
EmerinThe X chromosome EMD gene encodes the nuclear membrane protein emerin, a serine-rich member of the family of lamin-associated nuclear proteins. Mutations in EMD are associated with X-linked Emery-Dreifuss muscular dystrophy, which is characterized by early onset joint contractures appearing in childhood or adolescence; progressive limb weakness and atrophy; and cardiac involvement featuring conduction disorders, ventricular arrhythmias, and DCM. Sudden cardiac death in affected patients can be triggered by asystole and is therefore preventable by pacemaker implantation; however, in some patients there is a risk of sudden death from fibrosis related to the cardiomyopathy, thus requiring placement of an ICD.31,32 Female mutation carriers tend to have a less severe phenotype or do not develop the disease. Most disease-causing mutations are truncating, resulting in the total absence of normal emerin protein in the nucleus; however, DCM is also triggered by emerin mutations that alter a single amino acid. An interesting example is the Val26Ala mutation. This variant was identified in several index patients in a Spanish study assessing the presence of genetic alterations in patients undergoing heart transplantation for DCM. The identified patients underwent surgery at 2 hospitals in Madrid, and all came from a Canary Island population with an established history of DCM. These patients showed no evidence of skeletal myopathy or conduction disorders, which are frequently associated with other EMD mutations. Female carriers of the variant in heterozygosis showed no signs of cardiomyopathy.33
DCM CAUSED BY MUTATIONS IN DESMOSOME GENESDesmosomes are filament-linked protein plaques that maintain the structural integrity of contacts between adjacent cells. Mutations in desmosome genes are predominantly associated with the development of arrhythmogenic cardiomyopathy (ACM); however, there are forms of the disease of left ventricular or biventricular origin that are indistinguishable from DCM. The genes most frequently associated with this phenotype are DSP and PKP2, although associations have also been identified with other desmosome genes.34 For most genetic variants, the cardiomyopathy is autosomal dominant, but there are some recessive forms (such as Carvajal syndrome). Documented cases were characterized by an elevated incidence of ventricular arrhythmias and an increased risk of sudden cardiac death that was independent of ejection fraction.35 Left-dominant ACM is a frequently underdiagnosed condition due to symptom overlap with other cardiomyopathies and other conditions, such as myocarditis and idiopathic ventricular tachycardia. Left-dominant ACM should be suspected in patients with left ventricular arrhythmias and inferolateral T-wave inversion.36
GENES LESS FREQUENTLY ASSOCIATED WITH DCMRBM20RBM20 encodes a member of the SR protein family, named for the presence of a characteristic serine/arginine-rich domain. SR proteins regulate the alternative splicing of multiple genes, most notably TTN. The mutations in this gene identified to date are associated with autosomal dominant DCM. Most of them are missense mutations located in 2 functionally important regions, the serine/arginine-rich region (exon 9) and the zinc finger domain (exon 14), although other variants are distributed throughout the gene. Studies have revealed a poor prognosis in some patients, with high incidences of sudden cardiac death, heart failure, and heart transplantation.37,38 Other authors found no differences in survival or ventricular arrhythmia incidence between mutation carriers and noncarriers; however, the overall event rate in this study was low.39 The location of the mutation in the gene is posited to determine different disease subtypes, but this hypothesis remains unconfirmed.
FLNCFLNC encodes a cytoplasmic actin-binding protein. The first FLNC mutations identified are associated with skeletal myofibrillar myopathy, but recent data indicate that the main clinical phenotype related to FLNC mutations may be autosomal dominant cardiomyopathies. A multicenter study led by our group recently found that truncating FLNC mutations are associated with a highly penetrant overlapping phenotype between DCM and left-dominant ACM.40 The disease is characterized by almost exclusive left ventricular involvement, and notable disease features include dilatation and systolic dysfunction (sometimes mild in both cases), extensive areas of intramyocardial fibrosis in the left ventricular wall, a high frequency of ventricular arrhythmias, an absence of skeletal myopathy, and normal creatine kinase. The study found a high incidence of sudden cardiac death, especially in patients older than 40 years and even in patients with mild-to-moderate ventricular dysfunction.40 Recent evidence also links missense FLNC mutations to restrictive cardiomyopathy and HCM, although the evidence for these associations is less firm.
BAG3BAG3 encodes BAG family molecular chaperone regulator 3. This protein has antiapoptotic activity and is highly expressed in skeletal and cardiac muscle, where it is located in the Z-disc. Cardiovascular phenotypes associated with BAG3 have only recently been described clinically, and to date few mutations have been identified.41 Of the known BAG3 variants causing autosomal dominant DCM, most are exon deletions or truncating mutations, although a smaller number of disease-associated missense mutations has been identified; some of the identified mutations generate a severe phenotype.42 Some missense mutations are associated with myofibrillar myopathy, also with autosomal dominant inheritance.
PLNPLN encodes phospholamban, a protein located in the sarcoplasmic reticulum membrane. The p.Arg14del mutation in PLN is associated with DCM and has a founder effect in the Netherlands. This variant has been linked to the development of left-dominant ACM and arrhythmic forms of autosomal dominant DCM; mutation carriers are at high risk of malignant ventricular arrhythmias and advanced heart failure in young adulthood, with the rate of ventricular arrhythmias similar to that observed for LMNA mutations.43 A low amplitude R-wave has been identified as an early disease marker in mutation carriers and correlates with late gadolinium enhancement on cardiovascular magnetic resonance. Another study found low penetrance and milder phenotypes, especially for truncating mutations.44 Other PLN variants are related to the development of HCM.
SCN5ASCN5A encodes sodium voltage-gated channel alpha subunit 5, also known as Nav1.5. Most of the described mutations are associated with long QT and Brugada syndromes with an autosomal dominant inheritance pattern. Other phenotypes associated with SCN5A mutations are progressive conduction system disease (Lev-Lenègre disease), sinus node disease, and atrial fibrillation. A relatively small number of SCN5A mutations cause DCM; the best characterized is p.Arg222Gln, which cosegregates with the disease in multiple families from several countries.45 Most of the DCM-linked mutations are missense mutations located in the highly conserved transmembrane segments S3 and S4. The pathophysiological mechanism by which these variants cause DCM is unclear, but several hypotheses have been proposed.46
LAMP2LAMP2 encodes a lysosomal membrane protein, and mutations in this gene cause Danon disease, also known as glycogen storage disease type IIb. Several cardiomyopathy-associated mutations have been described that manifest very differently in men and women. Male patients (hemizygotes) develop HCM in childhood, and the disease features very severe hypertrophy and early progression to systolic dysfunction. In contrast, the initial diagnosis in women is usually DCM. Patients frequently show conduction alterations and pre-excitation on electrocardiography; moreover, associations have been reported with skeletal myopathy, retinopathy, and cognitive impairment, predominantly in men.47 Men have a worse prognosis and normally die between the ages of 30 and 40 years; however, the disease is also severe in women, in whom it is the form of DCM with the worst prognosis.
Mitochondrial genesMitochondrial diseases can be caused by mutations in mitochondrial DNA and thus show matrilineal inheritance. Nevertheless, most genes involved in the maintenance of mitochondrial structure and function are located in the nuclear genome and thus show Mendelian inheritance, usually with an autosomal recessive pattern. Mitochondrial disease should be suspected in patients with central nervous system disorders and multisystem disease. One of the most prominent mitochondrial diseases associated with DCM is Barth syndrome, which is caused by mutations in TAZ. The TAZ gene product tafazzin is involved in the mitochondrial metabolism of cardiolipin. Barth syndrome manifests as cardiomyopathy (dilated, hypertrophic, or with left ventricular noncompaction), endocardial fibroelastosis, skeletal myopathy, growth delay, neutropenia, and organic aciduria. Inheritance is X-linked, and women are usually asymptomatic carriers.48
Additional factors influencing familial DCMGiven the phenotypic variability and incomplete penetrance observed in DCM patients sharing the same genetic alteration, it is clear that disease manifestation and prognosis can be influenced by other factors, both favorably and unfavorably. These factors include additional genetic variants, as well as environmental and epigenetic factors.
Most DCM-associated genes (LMNA, RBM20, and sarcomere genes) cause disease predominantly in men.49 Moreover, some genes are associated with marked sex differences in disease progression and prognosis.50 Such differences are easy to explain for diseases caused by X chromosome genes, such as DMD and EMD; however, for genes located on other chromosomes, these differences currently lack a satisfactory explanation.
Physical exercise is generally recommended for heart failure patients, but for specific disease etiologies it can increase the risk of arrhythmia. For carriers of desmosomal gene mutations, resistance training has been found to increase disease penetrance and progression to clinical heart failure and arrhythmias.51 In HCM patients with mutations in sarcomere genes, intense physical activity is linked to earlier diagnosis, but there are no reported major prognostic differences.52 In carriers of disease-causing LMNA mutations, participation in competitive sports increases the risk of adverse events even if the activity ceased several years before diagnosis.53
Several studies have shown an influence of other environmental factors on the expression of familial DCM. These factors include myocarditis, nutritional deficiences, and cytotoxic agents.54–56 Alchohol consumption is regarded as an important etiological factor in DCM, and recent studies have shown an elevated genetic predisposition in patients with alcoholic DCM.57
CONCLUSIONS AND FUTURE PERSPECTIVES ON THE ROLE OF GENETICS IN DCM DIAGNOSIS AND RISK STRATIFICATIONIncreasing knowledge of the genetic basis of DCM has allowed the identification of a genetic cause in a high proportion of patients with diseases previously labeled idiopathic DCM. A comprehensive genetic test can identify 1 or several variants that explain the disease in approximately 50% of cases with no other identifiable cause, and this percentage exceeds 70% for familial disease. These advances furthermore confirm that idiopathic DCM is not a single disease, but rather a group of diseases, each with its distinct etiology, clinical course, and prognosis.
These findings increase the complexity of diagnostic approaches in patients with DCM, but at the same time constitute an essential advance for the development of individualized or personalized approaches to diagnosis, prognosis, and therapy.
Early diagnosis of mutation-carrying family members allows appropriate monitoring and measures to block disease progress. In affected patients, identification of the specific cause of DCM often provides information on the risk of disease progression and sudden cardiac death, with potentially important therapeutic implications.
Interpretation of the results of genetic studies is complex and requires multidisciplinary expertise encompassing molecular biology, genetics, and medical specialties (cardiology in the present context). It is not enough simply to identify one or several genetic variants associated with the disease; medical teams must instead integrate all available information about these variants in order to reach conclusions about the natural history of the different forms of disease produced. Achieving this goal is a major challenge due to the existence of multiple genetic variants in each of the many disease-associated genes, each with its specific consequences and severity of impact. To date, most studies have sought to simplify, limiting their scope to the comparison of patients with and without mutations or with mutations in different genes; however, this approach is inadequate.
Phenotype and risk depend on the identity of the mutated gene, but more precisely they depend on the nature of the specific mutation (including the mutation type, the region affected, etc) and the presence of other genetic or environmental factors. Almost all genes have variants associated with a higher or lower disease risk or that do not produce disease. It is therefore a mistake to limit discussion to low- and high-risk genes. Today it is possible to analyze the effect of individual variants, or at least groups of variants with shared characteristics and a similar functional impact. This information should be integrated with the available clinical information through a reevaluation of the diagnostic and prognostic criteria for the disease. As highlighted in this review, current clinical practice guidelines contain examples of how knowledge of a disease-causing mutation can radically alter the clinical management of affected patients. In the coming years, the number of such examples will grow, and it will be the norm to build diagnostic, prognostic, and therapeutic criteria on a foundation of knowledge about the specific natural history of the disease in question, whether the cause be monogenic, polygenic, or multifactorial. Progress in this direction will require extensive knowledge of the genetic and molecular basis of DCM and a specific analysis of the relationship between genetic variants, disease progression, and the response to distinct prevention and treatment strategies for each of the specific forms of the disease.
CONFLICTS OF INTERESTL. Monserrat is a shareholder in Health in Code S.L.
.