

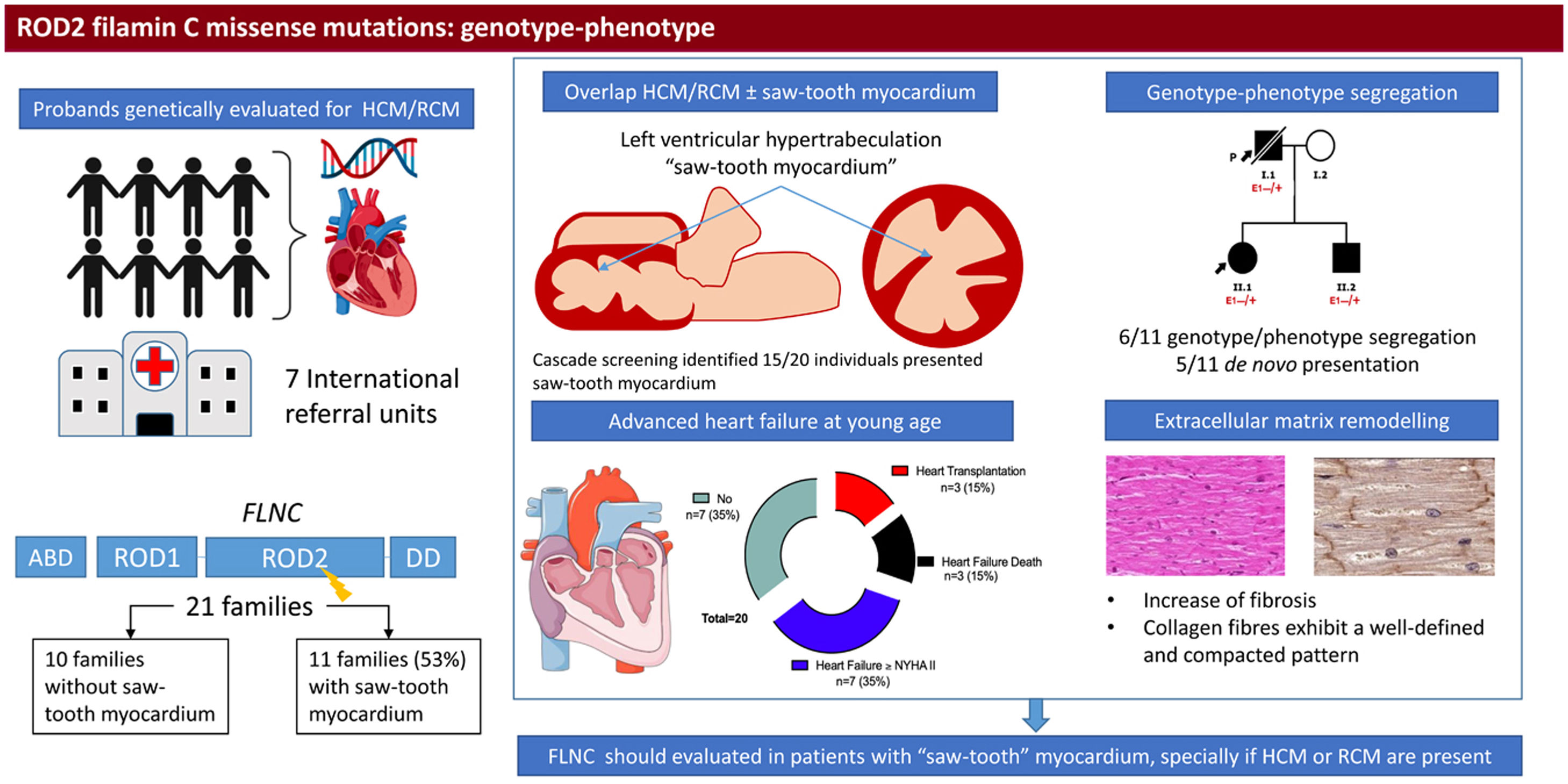
Missense mutations in the filamin C (FLNC) gene have been reported as cause of inherited cardiomyopathy. Knowledge of the pathogenicity and genotype-phenotype correlation remains scarce. Our aim was to describe a distinctive cardiac phenotype related to rare missense FLNC variants in the ROD2 domain.
MethodsWe recruited 21 unrelated families genetically evaluated because of hypertrophic cardiomyopathy (HCM)/restrictive cardiomyopathy (RCM) phenotype carrying rare missense variants in the ROD2 domain of FLNC (FLNC-mRod2). Carriers underwent advanced cardiac imaging and genetic cascade screening. Myocardial tissue from 3 explanted hearts of a missense FLNC carrier was histologically analyzed and compared with an FLNC-truncating variant heart sample and a healthy control. Plasmids independently containing 3 FLNC missense variants were transfected and analyzed using confocal microscopy.
ResultsEleven families (52%) with 20 assessed individuals (37 [23.7-52.7]) years showed 15 cases with a cardiac phenotype consisting of an overlap of HCM-RCM and left ventricular hypertrabeculation (saw-tooth appearance). During a median follow-up of 6.49 years, they presented with advanced heart failure: 16 (80%) diastolic dysfunction, 3 heart transplants, 3 heart failure deaths) and absence of cardiac conduction disturbances or skeletal myopathy. A total of 6 families had moderate genotype-phenotype segregation, and the remaining were de novo variants. Differential extracellular matrix remodeling and FLNC distribution among cardiomyocytes were confirmed on histology. HT1080 and H9c2 cells did not reveal cytoplasmic aggregation of mutant FLNC.
ConclusionsFLNC-mRod2 variants show a high prevalence of an overlapped phenotype comprising RCM, HCM and deep hypertrabeculation with saw-tooth appearance and distinctive cardiac histopathological remodeling.
Keywords
The increasing availability of next-generation sequencing technology has led to the important insight that cardiomyopathies are genetically heterogeneous disorders with different expressivity and penetrance. In most cardiomyopathies, the genotype-phenotype mechanisms within the same mutant gene remain widely unknown.
The FLNC gene encodes the main cardiac protein filamin C (FLNC), expressed in striated muscle cells, interacting with several proteins at Z-disk of the sarcomeres and with structural and signaling functions in the cardiomyocyte. The gene contains an actin-binding region, 2 hinge regions and a domain with 24 Ig-like repeats. FLNC plays a role in myogenesis, preserving myofibrils and sarcomere ultrastructure. Moreover, FLNC acts as an anchor membrane protein to the structural cytoskeletal proteins.1,2
Classically, FLNC mutations have been associated with a spectrum of inherited muscular diseases known as myofibrillar myopathies.3 In the last decade, a few reports based on a limited number of patients have described FLNC missense variants causing inherited restrictive cardiomyopathy (RCM),4–6 hypertrophic cardiomyopathy (HCM),7 or noncompaction cardiomyopathy (NCCM)8 with nearly absent skeletal muscle involvement. More recently, several studies have reported truncating variants in FLNC (FLNC-tv) related to arrhythmogenic cardiomyopathy.9,10 Despite being an increasingly recognized genetic cause of inherited cardiomyopathy, the causative relationship between FLNC missense variants and overlapped cardiac phenotypes is not clearly established.
Our aim was to describe phenotypic features of cardiomyopathy related to missense FLNC variants in the ROD2 domain (FLNC-mRod2).
METHODSStudy population and clinical evaluationWe collected all probands referred for cardiovascular genetic evaluation at the same reference laboratory because HCM or RCM showing an FLNC-mRod2 variant, and who were available for study. We excluded all individuals with another causative variant to explain the phenotype. After the first evaluation, we selected those families with saw-tooth appearance at cardiovascular imaging for an in-depth clinical study.
We performed a comprehensive clinical assessment including a detailed evaluation of the patient's clinical history, pedigree, creatine kinase, 12-lead electrocardiogram, Holter monitoring, and expert echocardiography assessment. Left ventricular ejection fraction dysfunction was assessed by the Simpson biplane method.11 Diastolic function was evaluated according to current recommendations.12 Left ventricle (LV) myocardium appearance was analyzed with a special focus on hypertrabeculation, according to current diagnostic criteria for NCCM with 2 well-defined layers (thin compacted and thick noncompacted layers).13 Saw-tooth myocardium was identified as the presence of multiple deep projections and invaginations in a dense and compacted myocardium.
The study was approved by the local ethics committee. Informed written consent for inclusion in the study was obtained from all participants or, in the case of minors or deceased individuals, from first-degree family members.
Genetic sequencingBlood samples from probands were obtained for genetic sequencing using a phenotype-guided next-generation sequencing panel at the same institution. In probands with an overlap or nondefined phenotype, we used a next-generation sequencing panel including genes related or candidates for cardiac disease. Coding exons and intronic boundaries of 247 genes related to inherited cardiovascular diseases () were captured using a custom probe library.
For the pathogenicity assessment of genetic variants, we considered information such as frequency in public databases (eg, the Human Gene Mutation Database the Single Nucleotide Polymorphism Database, NHLBI GO Exome Sequencing Project and ClinVar or in the Genome Aggregation Database,14–16 its previous description in the literature and preservation among species. After identification of rare FLNC variants in the index patients, we performed a genetic cascade screening among all available relatives using Sanger DNA sequencing. For the de novo variants, paternity was confirmed when available. Finally, the logarithm of the odds score17 and American College of Medical Genetics and Genomics (ACMG)18 classification were evaluated. For detailed methods regarding the genetic analysis see supplementary data.
Histological evaluationA complete histological, histochemical and immunohistochemical characterization of cardiac tissue belonging to 3 explanted hearts (patient from family F and family C, patients 2 and 3) was performed. Tissue samples were fixed for at least 48 hours in 10% neutral buffered formaldehyde, washed, dehydrated in ascending concentration of ethanol, cleared in xylol, and finally embedded in paraffin following a conventional protocol.19
Histological section of 5 μm thickness were obtained, dewaxed, hydrated and underwent the following steps: a) general histological examination was conducted with hematoxylin-eosin; b) the cardiomyocytes myofibrils were histochemical stained with Heidenhain hematoxylin; c) the organization and distribution of the main extracellular matrix (ECM) components were evaluated by Picrocirius collagen staining, Gomori's metal reduction technique for reticular fibers, Orcein histochemical staining for elastic fibers, and Alcian Blue staining for proteoglycans. In addition, collagens type I and IV (COL-I and COL-IV respectively) were identified by immunohistochemistry; d) the distribution pattern of FLNC and connexin-43 (Cx-43) were determined by immunohistochemistry. Technical details of the antibodies used are summarized in .
In addition to the histological characterization, a semiquantitative analysis of the cell/collagen ECM area ratio was carried out in 3 different sections from 3 different tissue slides stained with the Picrocirius technique by using Image J software (National Institute of Health, United States) and following a previously described procedure.20 Furthermore, 20 points were selected in 3 sections from each condition stained with the Heidenhain hematoxylin method and the intensity of the histochemical reaction for myofibrils was determined. Finally, the cell width was calculated in 20 cells per image selected in 3 images from each condition.
To detect specific histological changes attributable to FLNC-mRod2 variants, we compared the histological findings with a nonpathological heart tissue sample (control), and with an FLNC-truncating variant (FLNC-tv) carrier sample from our historical cohort. The FLNC-tv case showed the recognized overlapping phenotype of dilated cardiomyopathy and arrhythmogenic cardiomyopathy.9,10
Plasmid generation, cell culture, and confocal microscopyThe wild-type plasmid pCMV6-FLNC (212462; OriGene Technologies, United States) was used as a template to insert the variants p.P2301L, p.E3224K and p.R2340W using the Quik Change Lightning Kit (Agilent Technologies, United States) in combination with appropriate oligonucleotides (). The FLNC encoding complementary deoxyribonucleic acid (cDNA) of all plasmids was verified by Sanger sequencing (Macrogen, Netherlands). HT1080 (DSMZ, German Collection of Microorganisms and Cell Culture, ACC315, Germany) and H9C2 cells (ATCC, CRL-1446, United States) were cultured in μSlide chambers (ibidi GmbH, Germany) using Dulbecco's Modified Eagle Medium supplemented with 10% fetal calf serum and penicillin/streptomycin. Lipofectamin 3000 (Thermo Fisher Scientific, United States) was used for cell transfection according to the manufacturer's instructions. Twenty-four hours after transfection, cells were washed with phosphate buffered saline and were fixed for 10 minutes at room temperature using 4% Histofix (Carl-Roth, Germany). Immunocytochemistry analysis was performed as recently described using the TCS SP8 confocal microscope (Leica Microsystems, Germany).4
Statistical analysisStatistical analyses were conducted using SPSS Statistics software, version 21.0. (IBM Corp, United States). Continuous variables are reported as mean value±standard deviation for each measurement. For quantification of histological variables, we performed the Mann-Whitney U test (nonparametric). All categorical variables are expressed as frequencies and percentages.
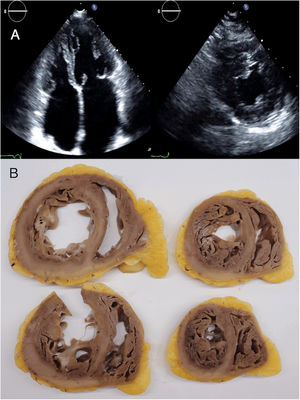
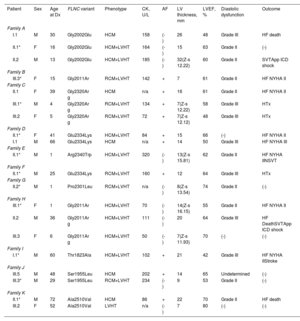
RESULTSPatients’ characteristicsIn total, we recruited 21 unrelated families from 7 reference centers who were genetically tested for HCM/RCM and found to carry rare FLNC-mRod2 variants (). A total of 11 families (52%) with 20 individuals (37 [23.7-52.7] years) showed 15 cases with a cardiac phenotype consisting of HCM or RCM with a marked and atypical left ventricular hypertrabeculation (LVHT) distorting LV with the presence of numerous cross bridging muscular projections. These protrusions of muscular bridges in the LV delineated deep crypts in a single, dense, and compacted myocardium (figure 1), which we have identified as “saw-tooth” appearance, different from the 2 characteristic layers of the NCCM (thin compacted and thick noncompacted layers). Furthermore, this singular LVHT was confirmed in 3 explanted hearts (figure 2).
 and echocardiogram (lower panel) images showing the saw-tooth left ventricular hypertrabeculation appearance with muscular projections and deep crypts distorting the normal left ventricular architecture. A, patient III.1 from family C; B, patient II.2 from family A; C, patient from family E.")
Cardiac magnetic resonance (top and center panels) and echocardiogram (lower panel) images showing the saw-tooth left ventricular hypertrabeculation appearance with muscular projections and deep crypts distorting the normal left ventricular architecture. A, patient III.1 from family C; B, patient II.2 from family A; C, patient from family E.
 and macroscopic images of explanted heart (B) from the index patient of family C, aged 24 and 36 years, respectively. Echocardiogram images showed left ventricular muscular protrusions. Pathological images showed the atypical and marked hypertrabeculation with profound recesses.")
Echocardiogram images (A) and macroscopic images of explanted heart (B) from the index patient of family C, aged 24 and 36 years, respectively. Echocardiogram images showed left ventricular muscular protrusions. Pathological images showed the atypical and marked hypertrabeculation with profound recesses.
Among the total cohort of 20 individuals, median age at diagnosis was 37 [23.7-52.7] years and 12 (60%) were male. HCM was more frequent (13; 65%) than RCM (4; 20%), and 15 individuals (75%) showed a saw-tooth appearance. A total of 16 individuals (80%) showed marked diastolic dysfunction (grade II or III) and 10 (50%) had atrial fibrillation, sometimes in adolescence or early adulthood.
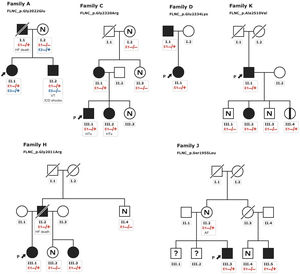
Patients HCM showed an increased interventricular septum (14.3±7 mm), a dilated left atrium (45.6±10.4 mm) and an initially preserved left ventricular ejection fraction (58.5±10.3%). A total of 2 individuals from family A and 2 from family C had bicuspid aortic valve. In addition, 9 patients underwent cardiac magnetic resonance with late gadolinium enhancement assessment, but none of them showed late gadolinium enhancement scar. All of them exhibited an abnormal electrocardiogram: 5 showed T wave inversion in precordial leads and 8 in inferior and precordial leads, 5 had right bundle branch block and 2 showed an unspecific intraventricular conduction delay. None of the patients had low voltages or atrioventricular block. Holter monitoring did not identify increased premature ventricular beats but 1 did identify nonsustained ventricular tachycardia. None of them had symptoms, signs, or a history of skeletal myopathy (mean creatine kinase level 137±58 U/L). Cascade genetic screening showed mild cosegregation of 6 variants with the phenotype (families A, C, D, H, J, K; figure 3), while the remaining 5 cases exhibited a de novo presentation (table 1, , ). Noncarriers showed manifestations of cardiomyopathy.
 indicates not available for evaluation. The arrows + P indicate the proband. Heterozygous carriers (E1 −/+) and noncarriers (E1 −/−). ICD, implantable cardioverter-defibrillator; HF, heart failure; HTx, heart transplant; VT, sustained ventricular tachycardia.")
Pedigrees of families with mild cosegregation. Squares indicate males, circles indicate females, slashes indicate deceased individuals, black shading indicates cardiomyopathy phenotype, black stripe indicates silent carrier, N indicates absence of phenotype, (?) indicates not available for evaluation. The arrows + P indicate the proband. Heterozygous carriers (E1 −/+) and noncarriers (E1 −/−). ICD, implantable cardioverter-defibrillator; HF, heart failure; HTx, heart transplant; VT, sustained ventricular tachycardia.
Clinical characteristics of evaluated carriers
| Patient | Sex | Age at Dx | FLNC variant | Phenotype | CK, U/L | AF | LV thickness, mm | LVEF, % | Diastolic dysfunction | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| Family A | ||||||||||
| I.1 | M | 30 | Gly2002Glu | HCM | 158 | (-) | 26 | 48 | Grade III | HF death |
| II.1* | F | 16 | Gly2002Glu | HCM+LVHT | 164 | (-) | 15 | 63 | Grade II | (-) |
| II.2 | M | 13 | Gly2002Glu | HCM+LVHT | 185 | (-) | 32(Z-s 12.22) | 60 | Grade II | SVTApp ICD shock |
| Family B | ||||||||||
| III.3* | F | 15 | Gly2011Ar | RCM+LVHT | 142 | + | 7 | 61 | Grade II | HF NYHA II |
| Family C | ||||||||||
| II.1 | F | 39 | Gly2320Ar g | HCM | n/a | + | 16 | 61 | Grade II | HF NYHA II |
| III.1* | M | 4 | Gly2320Ar g | RCM+LVHT | 134 | + | 7(Z-s 12.22) | 58 | Grade III | HTx |
| III.2 | F | 5 | Gly2320Ar g | RCM+LVHT | 72 | + | 7(Z-s 12.12) | 48 | Grade III | HTx |
| Family D | ||||||||||
| II.1* | F | 41 | Glu2334Lys | HCM+LVHT | 84 | + | 15 | 66 | (-) | HF NYHA II |
| I.1 | M | 66 | Glu2334Lys | HCM | n/a | + | 14 | 50 | Grade III | HF NYHA III |
| Family E | ||||||||||
| II.1* | M | 1 | Arg2340Trp | HCM+LVHT | 320 | (-) | 13(Z-s 15.81) | 62 | Grade II | HF NYHA IINSVT |
| Family F | ||||||||||
| II.1* | M | 25 | Glu2334Lys | RCM+LVHT | 160 | + | 12 | 64 | Grade III | HTx |
| Family G | ||||||||||
| II.2* | M | 1 | Pro2301Leu | RCM+LVHT | n/a | (-) | 8(Z-s 13.54) | 74 | Grade II | (-) |
| Family H | ||||||||||
| III.1* | F | 1 | Gly2011Ar | HCM+LVHT | 70 | (-) | 14(Z-s 16.15) | 55 | Grade II | HF NYHA II |
| II.2 | M | 36 | Gly2011Ar g | HCM+LVHT | 111 | (-) | 20 | 64 | Grade III | HF DeathSVTApp ICD shock |
| III.3 | F | 6 | Gly2011Ar g | HCM+LVHT | 50 | (-) | 7(Z-s 11.93) | 70 | (-) | (-) |
| Family I | ||||||||||
| I.1* | M | 60 | Thr1823Ala | HCM+LVHT | 102 | + | 21 | 42 | Grade III | HF NYHA IIStroke |
| Family J | ||||||||||
| III.5 | M | 48 | Ser1955Leu | HCM | 202 | + | 14 | 65 | Undetermined | (-) |
| III.3* | M | 29 | Ser1955Leu | RCM+LVHT | 234 | (-) | 9 | 53 | Grade II | (-) |
| Family K | ||||||||||
| II.1* | M | 72 | Ala2510Val | HCM | 86 | + | 22 | 70 | Grade II | HF death |
| III.2 | F | 52 | Ala2510Val | LVHT | n/a | (-) | 7 | 80 | (-) | (-) |
AF, atrial fibrillation; App, appropriate; CK, creatin kinase (reference values 11-145 U/L); dx, diagnostic; F, female; HCM, hypertrophic cardiomyopathy; HF, heart failure; HTx, heart transplant; ICD, implantable cardioverter-defibrillator; IVS, interventricular septum; LV, left ventricle; LVEF, left ventricular ejection fraction; LVHT, left ventricular hypertrabeculation; M, male; NSVT, nonsustained ventricular tachycardia; NYHA, New York Heart Association; RCM, restrictive cardiomyopathy; VT, sustained ventricular tachycardia; n/a, not available; (+), present; (-), not present.
The remaining 10 families identified with FLNC-mRod2 variants without the saw-tooth appearance consisted of 4 obstructive HCM (1 with LVHT but no saw-tooth trait) patients, 1 HCM on postmortem analysis, 1 apical HCM, 1 RCM and 3 individuals without cardiomyopathy.
EventsAfter a median follow-up of 6.49 [3.8-21.33] years after phenotype diagnosis, 11 (55%) patients showed symptomatic heart failure (HF) New York Heart Association (NYHA)≥II, with 3 of them requiring 3 a heart transplant due to advanced HF in their thirties. Three patients (15%) died from HF. Furthermore, 4 (26.6%) patients had syncope during follow-up and 2 had sustained ventricular tachycardia (SVT) and received an implantable cardioverter-defibrillator (ICD).
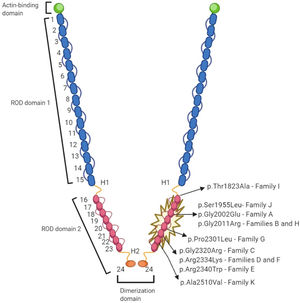
Genetic variantsFigure 4 represents the location of the 9 rare variants identified in the FLNC ROD2 domain. All the missense variants observed in this cohort were located in the ROD2 domain, specifically in 18 to 21 Ig-like domains. All identified variants were absent in control population databases and bioinformatics predictors showed that they were likely deleterious. ACMG classification identified 4 variants as likely pathogenic and 5 as variants of unknown significance (). A second relevant-causative genetic variant was not identified in any of the probands, and paternity was confirmed in 4 of the de novo cases. No CNVs were detected in the selected cohort. In all these cases, the quality of the sample was adequate to carry out this analysis.
Histology, histochemistry, and immunohistochemistry
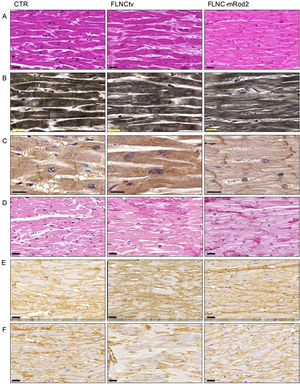
Hematoxylin-eosin staining revealed moderately larger cardiac cells in FLNC-tv heart samples compared with FLNC-mRod2 and controls (figure 5A). It also revealed a lower density of myofibrils and a slight decrease in intercellular space in FLNC-mRod2-affected tissues, resulting in a more compact tissue organization pattern in these specific samples (figure 5B). These results were confirmed by quantitative histological analyses (). The immunohistochemical analysis of FLNC confirmed the intracellular localization of this protein in all samples, but in FLNC-tv-affected samples, this protein was irregularly distributed and totally absent at the intercalated discs (ID) (figure 5C). In contrast, in FLNC-mRod2-affected tissues, it was present within the contractile apparatus, such as in the control group, but it was particularly positive at the ID level.
 showing the increase in the intercellular space in FLNC-tv and compaction in FLNC-mRod2. B, Heideinhain")
Histological pattern, myofibril histochemistry, FLNC immunohistochemistry and ECM remodeling analyses in FLNC genetically affected heart tissues. A, hematoxylin-eosin staining of control and genetically compromised heart tissue samples (FLNC-tv and FLNC-mRod2) showing the increase in the intercellular space in FLNC-tv and compaction in FLNC-mRod2. B, Heideinhain's hematoxylin for myofibrils demonstrating the minor density in FLNC-mRod2. C, Intracellular distribution of FLNC as determined by immunohistochemistry. Note the absence of FLNC in the intercalated disc of the FLNC-tv sample. D, Histochemical identification of fibrillar collagens by Picrosirius staining (in red) with an increase in these fibers in genetically affected samples. Immunohistochemical identification of collagen type I (E) and type IV (F). Scale bar: 20μm. CTR, control; FLNC-mRod2, missense variant in FLNC-Rod2 domain; FLNC-tv, filamin C truncating variant.
On the other hand, the—remodeling analysis revealed fibrotic changes in genetically affected samples vs the control (figure 5D), showing a clear increase in fibrillar collagen fibers. In FLNC-mRod2-affected samples, we observed this increase in collagen fibers around the cardiomyocytes, exhibiting a well-defined and compacted pattern, different to that observed for FLNC-tv (abundant and irregularly organized) (figure 5E-F). Finally, the fibrotic process was semiquantitatively confirmed, showing that the increased collagen content reached 49.2±7.9% in FLNC-tv followed by 29±2.5% in FLNC-mRod2 genetically affected samples, both being considerably higher than the 26.5±2.2% collagen content observed in the control (). Moreover, the remodeling of the collagen network—fibrotic response—was accompanied by an alteration of the reticular fibers and acid proteoglycans content, intensity, and pattern (). All these histological findings in FLNC-mRod2-affected samples were consistent in the 3 samples.
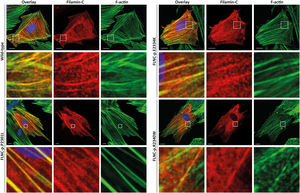
Functional analysesHT1080 and H9C2 cells were transfected with expression plasmids for wild-type and mutant FLNC (p.P2301L, p.E2334K and p.R2340W). Confocal analyses revealed a comparable cellular localization of wild-type and mutant FLNC in transfected cells (figure 6 and ) indicating that pathogenic protein aggregation was unlikely for the described missense variants.
. c-Myc tagged filamin C is shown in red. F-actin (green) was contained using phalloidin conjugated with Alexa-488. Representative cell images are shown. Scale bars = 10 μm.")
H9C2 cells were transfected with expression plasmids for wild-type and mutant filamin C (p.P2301L, p.E2334K and p.R2340W). c-Myc tagged filamin C is shown in red. F-actin (green) was contained using phalloidin conjugated with Alexa-488. Representative cell images are shown. Scale bars = 10 μm.
Recently, sporadic FLNC missense variants have been described associated with the development of RCM.4–6 It had also been postulated to be associated with HCM.7 Nevertheless, evidence comes from limited publications. Here we describe for the first time a distinctive cardiac phenotype in a multicenter cohort of patients with rare missense variants at the ROD2 domain of the FLNC gene, consisting of a severe HCM/RCM phenotype with an unusual saw-tooth LVHT. The HCM and RCM overlap has previously been described in the context of mutations in cardiac sarcomere protein genes.21 However, few cases of saw-tooth myocardium have been published to date and none have been associated with a specific gene.22 Interestingly, most of the patients exhibited the saw-tooth appearance on cardiovascular imaging and some exhibited it at young age. Although both saw-tooth myocardium and NCCM may be considered epiphenomena in the context of another condition, 3 isolated reports of individual patients have suggested a link between NCCM and missense mutations in FLNC.1,23,24 Although saw-tooth patients exhibited deep myocardial recesses, in all patients there was a single and compacted myocardial layer, rather than 2 distinct layers. Therefore, current diagnostic NCCM criteria were not applicable. Interestingly, Roldán-Sevilla et al.6 described an individual with the p.Pro2301Leu variant, like patient from family G, with RCM in the absence of LVHT. Although phenotypic description is succinct, this difference with our patient may be explained by the already known pleiotropic expression of FLNC.1
Regarding the clinical impact of FLNC-mRod2 variants, most individuals had HF symptoms and marked diastolic dysfunction. It is noticeable that in this small cohort of mostly young patients, there was a high incidence of major clinical events. This is in line with a more severe form of cardiomyopathy than classic HCM and similar to HCM due to mutations in thin filament protein genes such as troponin T.25 This finding might reveal that restrictive physiology and advanced HF in FLNC-mRod2 variants may be common at early stages of the disease, similarly to previous descriptions of patients with advanced HF at young ages.4,6 Interestingly, we observed a high incidence of de novo variants, which supports the likelihood of pathogenicity of the rare variants. Since paternity was confirmed in 4 variants, these are classified as likely pathogenic according to ACMG.18
In addition, we have shown evidence of a mild phenotype-genotype cosegregation in 4 independent families, a critical fact that reinforces pathogenicity. FLNC is a homodimeric protein encoded by the FLNC gene (7q32), composed by 47 coding exons. The protein is composed of an amino-terminal actin-binding domain and a ROD of 24 Ig-like domains which are connected by flexible hinge regions between domains 15 and 16 (hinge 1) and domains 23 and 24 (hinge 2).26 However, FLNC variants have been described as being spread all along the gene, and genotype-phenotype information and cardiac involvement is incomplete; previous reports have questioned the clinical severity, or even the pathogenicity, of common FLNC variants in classic forms of HCM, cases that are clearly different to those presented in our cohort27: all of our probands carried a missense mutation located at the same ROD2 subdomain, specifically affecting Ig-like domains from 18 to 21 (d18-21), and most of them shared this singular “saw-tooth myocardium” phenotype. The 2 domain pairs (18-19 and 20-21) are preserved in all vertebrate filamins.28 This ROD2 zone contains the domain responsible for the binding of membrane glycoproteins and it forms a semiflexible ROD with 2 hinge regions and 24 highly homologous tandem repeats, formed by 93 to 103 amino acid residues organized as Ig-type domains. Recent data exploring genotype-phenotype correlations among carriers of FLNC pathogenic variants suggest a cluster of missense variants in this region and the development of HCM phenotype.1 This d18-21 cluster interacts with Z-disc proteins, muscle development and contraction-related proteins. Furthermore, it is of special interest because it is a crucial point for protein phosphorylation.29 It is suggested that this ROD2 subdomain is essential for FLNC dimerization and secondary protein structure acquisition.23 Therefore, missense variants in the ROD2 subdomain may precipitate a misfolded protein and impaired crosslinking leading to sarcomere disarray and mechanotransduction impairment.23,30 This hypothesis should be confirmed in future studies.
Histologically, we characterized the tissue features of FLNC-mRod2 and FLNC-tv in heart samples from 3 explanted hearts and 1 case of sudden cardiac death case, respectively. Although they should be interpreted with caution due to the limited sample, the histological and histochemical analyses of the FLNC-mRod2 patient showed thin cells with a low density of myofibrils, which may be explained by an impaired crosslinking of the structural, contractile, or even anchor membrane-cytoskeletal proteins. Furthermore, we did not observe FLNC aggregates in histology or functional experiments. Here, as previously described,4,7 we used 2 heterologous systems, with the H9C2 cell line being more representative of myocyte physiology. Although abnormal protein has been observed within aggregates in the tissue of FLNC-associated HCM and RCM patients,31 Valdes-Mas7 and Brodehl4 have demonstrated that not all cardiomyopathy-associated FLNC missense variants induce protein aggregation, eg, the FLNC-mRod2 variants p.I2160F or p.V2297M.4,5 Interestingly, the absence of aggregates has been described in the cardiac tissue of patients with FLNC-tv in the ROD2 domain indicating the lack of an abnormal FLNC protein.23
We also confirmed the intracellular localization of FLNC in all heart samples, but there were differences among them. While FLNC-mRod2 showed an intense presence of FLNC in the ID, the FLNC-tv-affected samples expressed an irregularly distributed protein and was totally absent at the ID level. This absence of FLNC at the ID in FLNC-tv was previously reported in zebrafish models.31 Furthermore, we confirmed a different genotype-dependent ECM remodeling leading to diverse fibrotic changes in genetically affected patients. FLNC-tv-affected tissues were occupied by an abundant and irregularly organized fibrillar collagen network, confirming an evident fibrotic response.9 However, the FLNC-mRod2-affected samples exhibited a well-defined and compacted tissue pattern with a mild to moderate fibrotic process. This is in contrast with the late gadolinium enhancement results, but may be explained by diffuse myocardial fibrosis that cannot be evaluated by late gadolinium enhancement imaging. Future studies with T1 mapping would improve interstitial myocardial fibrosis evaluation.
LimitationsThe main limitation of this study is the limited number of patients evaluated and its retrospective design. A total of 5 of the 9 rare variants are considered variants of unknown significance, thus limiting the conclusions that can be drawn. For histology, given the nature of the samples, we included only a limited number of independent heart samples affected by this particular genetic condition, limiting the possibility of quantitative and statistical analyses. Cardiac magnetic resonance tissue characterization is limited by the retrospective nature of the study. Furthermore, HT1080 cells may not reproduce human cardiomyocyte physiology.
CONCLUSIONSRare missense FLNC variants in the ROD2 domain can exhibit an overlapping phenotype comprising HCM and RCM with a saw-tooth LVHT characterized by severe HF progression and distinctive cardiac histopathological remodeling (figure 7).

Central illustration. Identification and clinical features of patients with missense variants in the ROD2 domain of FLNC and saw-tooth myocardium. We included patients who were genetically evaluated due to hypertrophic cardiomyopathy/restrictive cardiomyopathy and with a missense variant in the ROD2 domain of the FLNC gene. Eleven probands exhibited saw-tooth myocardium. Cascade screening identified up to 15 individuals with this trait. In 6 families, we observed mild cosegregation. Patients developed heart failure at a young age. Pathology demonstrated distinct cardiac remodeling. FLNC, filamin C; HCM, hypertrophic cardiomyopathy; NYHA, New York Heart Association; RCM, restrictive cardiomyopathy.
A. Brodehl is grateful for the financial support of Ruhr-University Bochum (FoRUM, FoRUM-F937R2-2020). Dr Bermúdez Jiménez received funding support by a research training program Río Hortega by Carlos III Institute of Health (CM19/00227).
AUTHORS’ CONTRIBUTIONSAll authors made a significant contribution to this work by collecting the cases in their referral centers. F. J. Bermúdez-Jiménez and V. Carriel wrote and coordinated the manuscript equally. AB conducted the functional study. J. Jiménez-Jáimez supervised the clinical and research work and the final version of the manuscript. All authors approved the final version of the manuscript
CONFLICTS OF INTERESTNone.
AcknowledgmentsWe would like to thank the patients and their families for their generous and unconditional collaboration. We also thank Fabiola Bermejo Casares, Paloma de la Cueva Batanero (Department of Histology, University of Granada, Spain), Caroline Stanasiuk (EHKI, Germany), Raúl Franco Gutiérrez (University Hospital Lucus Augusti, Santiago de Compostela, Spain), Neus Baena Díez (University Hospital Parc Tauli, Sabadell, Spain), Carlos Gómez Navarro (University Hospital Torrecardenas, Almería, Spain) and Huafrin Kotwal (St. Bartholomew's Hospital, London, UK) for technical assistance. Histological analyses were supported by the Tissue Engineering Group (CTS-115), University of Granada, Spain.
- -
FLNC is a striated muscle protein encoded by FLNC that serves as a signaling and scaffolding protein.
- -
FLNC gene variants have been strongly associated with arrhythmogenic dilated cardiomyopathy and sporadically with hypertrophic cardiomyopathy, restrictive cardiomyopathy, and noncompaction cardiomyopathy.
- -
-This study provides new insight into the spectrum of FLNC cardiomyopathy and for the first time describes a novel association with an overlap cardiac phenotype comprising saw-tooth myocardium, hypertrophic and restrictive cardiomyopathy.
- -
This is the first study to associate a genetic basis with the saw-tooth myocardium pattern.
- -
The FLNC gene should be evaluated among patients with this nonspecific but severe cardiac phenotype for appropriate diagnosis and identification of relatives at risk.
Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1016/j.rec.2022.08.002