Sudden cardiac death (SCD) in young people often has a genetic cause. Consequently, the results of “molecular autopsy” may have important implications for their relatives. Our objective was to evaluate the diagnostic yield of a molecular autopsy program using next-generation sequencing.
MethodsWe performed a prospective study of a cohort of consecutive patients who died from nonviolent SCD, aged ≤ 50 years, and who underwent molecular autopsy using large panels of next-generation sequencing, with subsequent clinical and genetic family screening. We analyzed demographic, clinical, toxicological, and genetic data.
ResultsWe studied 123 consecutive cases of SCD in persons aged ≤ 50 years. The incidence of SCD was 5.8 cases/100 000 individuals/y, mean age was 36.15±12.7 years, and 95 were men (77%). The cause was cardiac in 53%, unexplained SCD in 24%, toxic in 10.6%, and infant SCD in 4%. Among cardiac causes, ischemic heart disease accounted for 38% of deaths, arrhythmogenic cardiomyopathy for 7%, hypertrophic cardiomyopathy for 5%, and idiopathic left ventricular hypertrophy for 11%. Genetic analysis was performed in 62 cases (50.4%). Genetic variants were found in 42 cases (67.7%), with a mean of 3.4±4 genetic variants/patient, and the variant found was considered to be pathogenic or probably pathogenic in 30.6%. In unexplained SCD, 70% showed some genetic variant. Family screening diagnosed 21 carriers or affected individuals, 5 of whom were at risk, indicating an implantable cardiac defibrillator.
ConclusionsProtocol-based and exhaustive study of SCD from cardiac causes in persons aged ≤ 50 years is feasible and necessary. In a high percentage of cases, the cause is genetic, indicating the existence of relatives at risk who could benefit from early diagnosis and treatment to avoid complications.
Keywords
Sudden cardiac death (SCD) is defined as the unexpected death of a person without any condition that would appear fatal within the first hour after symptom onset or when the deceased was last seen in apparently good health within the preceding 24h.1 SCD normally has a cardiac origin2 and is the leading cause of death in the industrialized world, with a prevalence of 20%. 3 SCD has a devastating psychosocial impact on family members of the deceased and, through the media, on society at large, especially when the victim is young. SCD of persons aged <35-40 years is in most cases due to heart disease with an underlying genetic cause (cardiomyopathy or channelopathy), indicating that the deceased's family members may also be at risk; in contrast, SCD of older individuals is usually caused by ischemic heart disease.2,4
The study of cases of SCD usually falls within the domain of forensic medicine.5,6
The combination of a complete cardiac histological examination and a histopathology-guided genetic analysis is known as molecular autopsy, and this procedure can identify the cause of SCD in a high proportion of individuals.7,8 Therefore, after the death of a young person, in addition to the mandatory forensic autopsy, it is advisable to perform a molecular autopsy in cases of suspected cardiomyopathy or when clinical autopsy detects no abnormalities or yields inconclusive findings.8 Unfortunately, this recommendation is seldom followed, either in Spain or in other countries.5,9
The term sudden unexplained death syndrome (SUDS) is applied to SCD of uncertain cause despite suitable autopsy and toxicological anlaysis.10 SUDS accounts for approximately 40% of SCDs of persons between the ages of 1 and 35-40 years.6,11.12 An estimated 10% to 25% of unexplained SCDs in adults and up to a third in infants and children can be attributed to channelopathies (long QT syndrome, short QT syndrome, Brugada syndrome, or catecholaminergic polymorphic ventricular tachycardia).2,12,14 In these cases, the diagnostic yield (defined as the additional benefit obtained) of genetic analysis by next-generation sequencing (NGS) is between 29% and 32%.15,16
Numerous published series have reported widely differing figures for the annual incidence of SCD, in part due to the different age ranges analyzed. For deaths between the ages of 1 and 35-40 years, the estimated incidence ranges from 1.3 to 8.5 cases/100 000 individuals/y.6,11,17–19
The goal of the present study was to assess the diagnostic yield of a sequential battery of tests in the diagnosis of SCD in young people. In addition to clinical, histological, and toxicological analyses, the test battery included genetic analysis with the largest NGS panels available followed by a clinical and genetic family cascade screening.
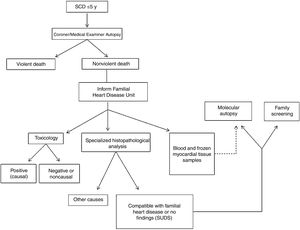
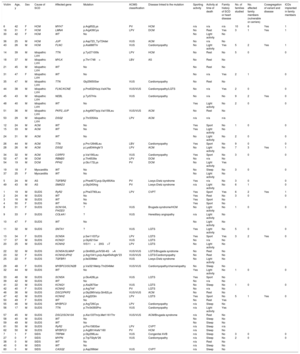
METHODSIn the Balearic Islands, hospitals and the regional Medical Examiner's Office (Instituto de Medicina Legal) has designed a collaborative and multidisciplinary program for the complete study of SCD in young people. The program is called MUSIB (from the Spanish title MUerte Súbita Islas Baleares; Sudden Cardiac Death in the Balearic Islands) and has been running continuously since February 2015. The Medical Examiner's Office holds records of all SCD cases in the Balearic Islands, a Spanish autonomous community with a population of 1 144 392 inhabitants (Spanish National Statistics Institute, 2016). To standardize the study of nonviolent SCD of persons aged ≤ 50 years, a working group was established that includes cardiologists, forensic scientists, pathologists, intensive care specialists, geneticists, biologists, and chemists. The working group established a protocol for diagnosing the cause of SCD that includes the performance of a full autopsy in all cases, including exhaustive pathological anatomy and genetic analysis.
The forensic autopsies reported here were performed by the Balearic Islands Medical Examiner's Office according to standardized protocols established in specialist guidelines.8 The heart and other internal organs were sent to the National Institute of Toxicology and Forensic Sciences in Barcelona for macroscopic, microscopic, and toxicological analysis. Blood samples were sent to the genetics laboratory at Son Espases University Hospital for later molecular analysis guided by the histopathology findings. Analyses were coordinated by the Familial Heart Disease Unit at Son Llàtzer Univeristy Hospital (figure 1).
; SCD, sudden cardiac death; SUDS, sudden unexplained death syndrome.")
As per the MUSIB protocol, we collected clinical and family histories, activity at time of death, electrocardiogram records if available, previous treatments, and the macroscopic and microscopic histopathology and toxicology results. An NGS genetic analysis was conducted when the histopathology revealed cardiomyopathy, yielded inconclusive findings, or detected no macroscopic or microscopic evidence of structural heart disease (thus indicating SUDS). Detected cardiomyopathies were studied with large panels of cardiomyopathy-related genes. When the histopathology was inconclusive or indicated SUDS, the NGS panel contained between 194 and 380 genes. Until 2017, NGS analysis was outsourced to laboratories run by Health in Code (A Coruña), Imegen (Valencia), and Centogene (Germany). Since 2017, samples have been analyzed in the genetics laboratory at Son Espases University Hospital. Sequence variants were sorted using a pre-established prioritization protocol based on the likely functional impact of the encoded protein and the variant's allelic frequency. The NGS analysis was supported by in silico prediction software tools. The pathogenicity of all genetic variants was assessed according to current criteria established by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP); the analysis excluded benign and likely benign variants and variants of uncertain significance (VUS) with a frequency ≥ 0.02% in the GnomAD, ClinVar, or Exac databases or with no demonstrated cosegregation in published studies.20 For rare genetic variants, findings were confirmed by Sanger sequencing. Because the protocol involves clinical and genetic family cascade screening (beginning with all first degree relatives and, when carriers are found, proceeding to second and third degree relatives, etc.), the cosegregation of the identified variants was studied whenever possible.
Data confidentiality was protected according to current regulations. The MUSIB program was approved by the Balearic Islands Ethics Committee.
Statistical analysisIn this descriptive analysis, qualitative variables are expressed as frequencies, and quantitative variables are expressed as mean±standard deviation or median [interquartile range]. Statistical differences were analyzed with the statistical package SPSS 15.0.
RESULTSTo date, the MUSIB program has examined 183 cases of SCD. Deaths lacking full test results at the time of the current analysis were excluded, leaving 123 consecutive cases of SCD with completed histopathological, chemical, and genetic analysis. This corresponds to an SCD incidence in the Balearic Island population aged ≤ 50 years of 5.8/100 000 individuals/y. The incidence was much higher among men (10.1/100 000 individuals/y, vs 1.6/100 000 individuals/y for women). Mean age at death was 36.15±12.7 (interval, 0-50) years; 95 victims were men (77%) and 28 were women (23%). The mean age of male victims was 37.5%±11.4 years, and the mean age of female victims was 31.5±15.6 years.
SCD prevalence varied with age group. Most deaths occurred among people aged 41 to 50 years (58 deaths; 47%). There were 32 deaths (26%) of persons aged 31 to 40 years, 20 (16%) of persons aged 21 to 30 years, 6 (4.9%) of persons aged 11 to 20 years, and 7 (5.7%) of persons aged 10 years or less, of whom 5 were infants. The number of male victims was higher in all groups except for infants.
The most frequent activity at time of death was rest (28.5%), followed by light or routine daily activity (25.2%), sleep (15.4%), and sporting activity (12.2%). Activity at time of death was unknown for 23 people (18.7%). Only 13 deaths (10.6%) were of people with a known family history of SCD or cardiomyopathy. Advanced cardiopulmonary resuscitation was attempted in 65 individuals (52.8%). Of the sample, 22 (17.9%) regularly took part in sporting activities (as amateurs or professionals). Within this subgroup, 13 (59%) died while exercising, 5 (22.7%) during light daily activity, and 3 (13.6%) while resting or sleeping.
ToxicologyMedicinal or recreational drug use was detected in 51.4% of autopsies but was the clear cause of SCD in only 13 individuals (10.6%). Among individuals with evidence of drug use, alcohol was detected in 69%, cocaine in 13%, cannabis in 8%, 6-monoacetylmorphine in 5%, and 3,4-methylenedioxymethamphetamine in 2%. Medicinal drugs (mostly benzodiazepines) were frequently detected, but mostly at subtoxic concentrations.
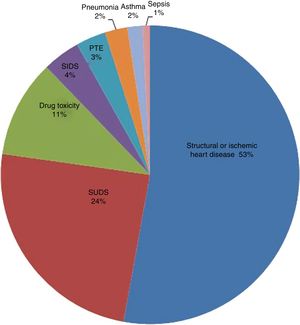
HistopathologyHistopathological analysis identified a cardiac cause in 95 deaths: 65 individuals (52.8%) with a structural or ischemic heart disease and 30 (24.4%) with no overt evidence of cardiomyopathy (indicating SUDS). Of the remaining deaths, 13 (10.6%) were due to drug toxicity, and less frequent causes were sudden infant death syndrome (SIDS) (4.1%), pulmonary thromboembolism (3.2%), pneumonia (2.4%), asthma (1.6%), and sepsis (0.8%) (figure 2).
; PTE, pulmonary thromboembolism; SIDS, sudden infant death syndrome; SUDS, sudden unexplained death syndrome.")
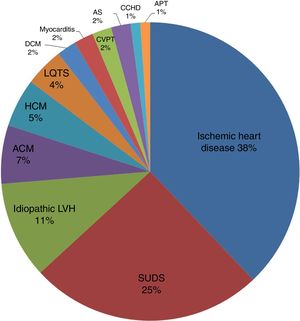
A breakdown of SCD cases with a cardiac cause (n=95, positive histopathology+SUDS) showed that 36 deaths (38%) were due to ischemic heart disease, 6 (7%) to arrhythmogenic cardiomyopathy, 5 (5%) to hypertrophic cardiomyopathy, 10 (11%) to idiopathic left ventricular hypertrophy, and 30 (31%) to SUDS. The figure for SUDS was later revised to 25% because molecular autopsy diagnosed long QT syndrome in 4 patients and polymorphic catecholaminergic polymorphic ventricular tachycardia in another 2. The remaining, less frequent causes of SCD were dilated cardiomyopathy (2 deaths 2%), myocarditis (2 deaths; 2%), acute aortic syndrome (2 deaths; 2%), complex congenital heart disease with atrioventricular block (1 death; 1%), and acute cardiac tamponade (1 death; 1%). The causes of SCD in the study cohort are summarized in figure 3.
. ACM, arrhythmogenic cardiomyopathy; APT, acute pericardial tamponade; AS, aortic syndrome; CCHD, complex congenital heart disease with atrioventricular block; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; LVH, left ventricular hypertrophy; SUDS, sudden unexplained death syndrome.")
Cardiac causes of SCD (n=95, positive histopathology+SUDS). ACM, arrhythmogenic cardiomyopathy; APT, acute pericardial tamponade; AS, aortic syndrome; CCHD, complex congenital heart disease with atrioventricular block; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; LVH, left ventricular hypertrophy; SUDS, sudden unexplained death syndrome.
There were 30 incidences of SUDS (66.7% men), occurring at a mean age of 36.7±10.9 years. Of these deceased persons, 30% had been active participants in sports. Of the people in this subgroup, 23.3% died in their sleep, 20% while at rest, 23.3% while performing light daily activities, and 23.3% while practicing sport. Overall, 13.3% of SUDS victims had a family history of sudden death or cardiomyopathy.
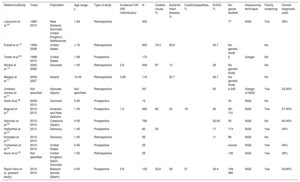
GeneticsA genetic analysis was performed in 62 individuals (50.4%) (table 1). This number included those who died from a nonischemic cardiac cause (excluding individuals who died from congenital heart disease and tamponade), together with the 5 infants who died of SIDS. A mean number of 3.4±4 genetic variants per person were found in a total of 42 individuals (67.7%). In 40 individuals, ACMG –AMP criteria classified the variants as VUS, pathogenic variants (PV), or likely pathogenic variants (LPV), representing a diagnostic yield of 64.5%. Considering only P and LP variants, the diagnostic yield was 30.6%.
SCD victims with a completed genetic analysis: clinical, genetic, and family data, ordered by cause of SCD
| Victim | Age, y | Sex | Cause of SCD | Affected gene | Mutation | ACMG classification | Disease linked to the mutation | Sporting activity | Activity at time of death | Family history of SCD or heart disease | No. of families studied | No. affected family members (vulnerable or carriers) | Cosegregation of variant and disease | ICDs implanted in family members |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 42 | F | HCM | MYH7 | p.Arg652Lys | PV | HCM | n/a | n/a | n/a | 10 | 6 | Yes | 1 |
| 16 | 31 | F | HCM | LMNA | p.Arg439Cys | LPV | DCM | No | Rest | Yes | 3 | 1 | Yes | 1 |
| 30 | 42 | F | HCM | WT | No | No | Light activity | No | ||||||
| 36 | 25 | M | HCM | JUP | p.Asp723_Tyr724del | VUS | ACM | No | n/a | No | ||||
| 43 | 26 | M | HCM | FLNC | p.Ala688Thr | VUS | Cardiomyopathy | No | Light activity | Yes | 5 | 2 | Yes | 1 |
| 14 | 39 | M | Idiopathic LVH | TTN | p.Tyr27100fs | LPV | HCM | No | Rest | No | 5 | 0 | 0 | |
| 18 | 37 | M | Idiopathic LVH | MYLK | p.Thr1748= | LBV | AS | No | Rest | No | ||||
| 21 | 45 | M | Idiopathic LVH | WT | No | No | Rest | No | ||||||
| 31 | 47 | F | Idiopathic LVH | WT | No | No | n/a | Yes | 2 | 1 | 0 | |||
| 35 | 47 | M | Idiopathic LVH | TTN | Gly25650Ser | VUS | Cardiomyopathy | No | Rest | No | ||||
| 44 | 38 | M | Idiopathic LVH | FLNC/KCNE | p.Pro632His/p.Val47Ile | VUS/VUS | Cardiomyopathy/LQTS | No | n/a | Yes | 2 | 0 | 0 | |
| 45 | 43 | M | Idiopathic LVH | NEBL | p.Tyr57His | VUS | Cardiomyopathy | No | n/a | No | 9 | 2 | Yes | 0 |
| 48 | 40 | M | Idiopathic LVH | WT | No | Yes | Light activity | No | 2 | 0 | 0 | |||
| 51 | 36 | M | Idiopathic LVH | PKP2, JUP | p.Arg490Trp/p.Val159Leu | VUS/VUS | ACM | No | Rest | No | ||||
| 53 | 29 | M | Idiopathic LVH | DSG2 | p.Thr335Ala | LPV | ACM | n/a | n/a | n/a | ||||
| 12 | 24 | M | ACM | WT | No | Yes | Sport | No | 1 | 0 | 0 | |||
| 15 | 33 | M | ACM | WT | No | Yes | Light activity | No | ||||||
| 24 | 31 | M | ACM | WT | No | No | Light activity | No | 2 | 0 | 0 | |||
| 26 | 44 | M | ACM | TTN | p.Pro12648Leu | LBV | Cardiomyopathy | Yes | Sport | No | 9 | 0 | 0 | |
| 28 | 26 | M | ACM | DSG2 | p.Lys834Argfs*3 | LPV | ACM | No | Light activity | No | 7 | 3 | Yes | 1 |
| 34 | 32 | M | ACM | CSRP3 | p.Val190Leu | VUS | Cardiomyopathy | Yes | Sport | No | 3 | 0 | 0 | |
| 52 | 47 | M | DCM | RBM20 | p.Thr653Ile | LPV | DCM | No | n/a | No | ||||
| 54 | 19 | M | DCM | PPA2 | p.Glu172Lys | PV | DCM | No | Light activity | Yes | ||||
| 19 | 18 | F | Myocarditis | WT | No | No | Sleep | No | 3 | 0 | 0 | |||
| 37 | 25 | F | Myocarditis | WT | No | No | Light activity | No | ||||||
| 5 | 24 | M | AS | TGFBR2 | p.Phe467Cys/p.Gly490Ala | PV | Loeys-Dietz syndrome | Yes | n/a | No | 3 | 0 | 0 | |
| 49 | 43 | M | AS | SMAD3 | p.Gly245Arg | PV | Loeys-Dietz syndrome | n/a | Light activity | No | 6 | 1 | 0 | |
| 1 | 19 | M | SUDS | RyR2 | p.Phe3790Leu | LPV | CVPT | Yes | Sport | Yes | 6 | 2 | Yes | 1 |
| 2 | 24 | M | SUDS | WT | No | Yes | Rest | No | 3 | 0 | 0 | |||
| 3 | 16 | M | SUDS | WT | No | Yes | Sport | No | ||||||
| 4 | 50 | F | SUDS | WT | No | Yes | Sport | No | ||||||
| 8 | 31 | F | SUDS | SCN10A, FHOD3 | ? | VUS | Brugada syndrome/HCM | No | Light activity | No | 1 | 0 | 0 | |
| 9 | 33 | F | SUDS | COL4A1 | ? | VUS | Hereditary angiopathy | n/a | Light activity | No | ||||
| 10 | 47 | F | SUDS | WT | No | No | Light activity | No | ||||||
| 11 | 32 | M | SUDS | SNTA1 | ? | VUS | LQTS | Yes | Light activity | No | 5 | 0 | 0 | |
| 13 | 34 | F | SUDS | SCN5A | p.Ser1103Tyr | LPV | LQTS | Yes | Sport | Yes | 3 | 2 | Yes | 0 |
| 17 | 37 | M | SUDS | KCNQ1 | p.Gly621Ser | VUS | LQTS | No | n/a | No | ||||
| 20 | 25 | M | SUDS | KCNH2 | IVS11+20G>T | LPV | LQTS | No | Light activity | No | ||||
| 22 | 32 | M | SUDS | SCN5A/SLMAP | p.Gln692Lys/IVS8-4G>A | VUS/VUS | LQTS/Brugada syndrome | No | Rest | No | ||||
| 23 | 32 | F | SUDS | KCNH2/JPH2 | p.Arg1041Lys/p.Asp454Argfs*23 | VUS/VUS | LQTS/Cardiomyopathy | No | Rest | No | ||||
| 25 | 22 | F | SUDS | TGFBR1 | p.Ile339Met | VUS | Loeys-Dietz syndrome | No | Light activity | No | ||||
| 29 | 48 | F | SUDS | MYBPC3/SCN2B | p.Val321Met/p.Thr204Met | VUS/VUS | Cardiomyopathy/channelopathy | No | Sleep | No | ||||
| 32 | 44 | M | SUDS | WT | No | Yes | Light activity | No | 1 | 0 | 0 | |||
| 33 | 48 | M | SUDS | SCN5A | p.Glu428Lys | VUS | LQTS | Yes | Sport | No | ||||
| 39 | 42 | M | SUDS | WT | No | No | n/a | No | ||||||
| 41 | 22 | M | SUDS | KCNQ1 | p.Ala287Ser | VUS | LQTS | No | Sleep | No | 1 | 0 | 0 | |
| 42 | 40 | F | SUDS | KCNH2 | p.Arg744* | PV | LQTS | No | n/a | No | ||||
| 46 | 36 | M | SUDS | DSC2/PKP2 | p.Gly286Val/p.Gln62Lys | VUS/VUS | ACM | No | Rest | No | ||||
| 47 | 42 | M | SUDS | KCNH2 | p.Arg22Gln | LPV | LQTS | Yes | Sport | No | 5 | 1 | Yes | 0 |
| 50 | 49 | F | SUDS | WT | No | No | Rest | Yes | ||||||
| 55 | 46 | M | SUDS | MYBPC3 | p.Arg726Cys | LPV | Cardiomyopathy | n/a | Sleep | No | ||||
| 56 | 17 | M | SUDS | TTN | p.Thr34393Pro | VUS | Cardiomyopathy | n/a | Light activity | Yes | ||||
| 57 | 45 | M | SUDS | DSC2/SCN10A | p.Ala133Thr/p.Met1161Thr | VUS/VUS | ACM/Brugada syndrome | n/a | Rest | No | ||||
| 58 | 40 | M | SUDS | WT | No | No | Sleep | No | ||||||
| 59 | 49 | M | SUDS | WT | No | No | Sleep | No | ||||||
| 61 | 50 | M | SUDS | RyR2 | p.Pro1583Ser | LPV | CVPT | n/a | Sleep | n/a | ||||
| 62 | 50 | M | SUDS | MYBPC3 | p.Arg891Alafs*160 | PV | HCM | n/a | Sleep | No | ||||
| 7 | 1 | F | SIDS | TRPM4 | p.Gly298Leu | VUS | Congenital AVB | n/a | Sleep | No | 3 | 0 | 0 | |
| 27 | 0 | F | SIDS | MYPN | p.Trp7Glyfs*26 | VUS | Cardiomyopathy | n/a | Sleep | No | 2 | 0 | 0 | |
| 38 | 0 | M | SIDS | WT | No | n/a | Rest | No | ||||||
| 40 | 0 | M | SIDS | WT | No | n/a | Sleep | No | ||||||
| 60 | 0 | M | SIDS | CASQ2 | p.Asp398del | VUS | CVPT | n/a | Sleep | No |
ACM, arrhythmogenic cardiomyopathy; ACMG, American College of Medical Genetics and Genomics; AS, aortic syndrome; AVB, atrioventricular block; CVPT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; F, female; HCM, hypertrophic cardiomyopathy; ICD, implantable converter defibrillator; LPV, likely pathogenic variant; LQTS, long QT syndrome; LVH, left ventricular hypertrophy; M, male; PBV, likely benign variant; PV, pathogenic variant; SCD, sudden cardiac death; n/a, information not available; SIDS, sudden infant death syndrome; SUDS, sudden unexplained death syndrome; VUS, variant of unknown significance; WT, wild-type (no mutations).
The results of the genetic analysis among those who died of cardiomyopathies were as follows. Genetic variants were found in 2 of the 6 persons who died of arrhythmic cardiomyopathy, in 4 of the 5 who died of hypertrophic cardiomyopathy, in 6 of the 10 who died of idiopathic ventricular hypertrophy, and in both individuals who died of dilated cardiomyopathy (1 variant in each) (table 1).
Genetic tests were performed on all individuals with an autopsy diagnosis of SUDS. VUS, PV, or LPV were detected in samples from 21 individuals (70%). Of the detected variants, 15 were linked to channelopathies (10 to long QT syndrome, 3 to Brugada syndrome, and 2 to catecholaminergic polymorphic ventricular tachycardia) and 10 to structural genes (table 1). Considering only P and LP variants, the diagnostic yield was 26.6%.
Of the 5 cases of SIDS, 4 occurred during sleep. Genetic variants were identified in 3 infants, all of them VUS; 2 were linked to ion channels (1 to catecholaminergic ventricular tachycardia and 1 to atrioventricular block) and 1 to cardiomyopathy (table 1).
Family screeningThe MUSIB protocol indicates a clinical and genetic screening of the relatives of all deceased individuals analyzed by NGS. In the present analysis, 104 individuals from 26 families were assessed (a mean of 4 individuals per family). To date, 21 individuals have been diagnosed as carriers or affected by the condition. Based on stratified risk, 5 relatives of deceased individuals have been fitted with an implantable converter defibrillator (ICD) as a primary prevention measure. Of these patients, 3 have hypertrophic cardiomyopathies, 1 has an arrhythmogenic cardiomyopathy, and 1 has catecholaminergic ventricular tachycardia. One patient has had an appropriate discharge.
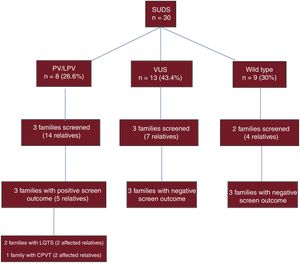
Studies have been performed in 25 relatives of individuals with an autopsy diagnosis of SUDS. To date, 3 individuals have been diagnosed with long QT syndrome and 2 with catecholaminergic polymorphic ventricular tachycardia (figure 4).

Diagnostic yield of genetic analysis in SCD victims with an autopsy diagnosis of SUDS and family screening in surviving relatives. CPVT, catecholaminergic polymorphic ventricular tachycardia; LPV, likely pathogenic variant; LQTS, long QT syndrome; PV, pathogenic variant; SCD, sudden cardiac death; SUDS, sudden unexplained death syndrome; VUS, variant of uncertain significance.
The family screening demonstrated the cosegregation of several genetic variants, confirming their pathogenicity. The variants showing cosegregation included the p.Ser1103Tyr variant of the SCN5A gene, an LPV that causes long QT syndrome; the p.Phe3790Leu variant of RyR2, an LPV that causes catecholaminergic polymorphic ventricular tachycardia; the p.Arg652Lys variant of MHY7, a PV that causes hypertrophic cardiomyopathy; and the p.Lys834Argfs*3 variant of DSG2, an LPV that causes arrhythmogenic cardiomyopathy. Family screening also detected possible cosegregation (only 1 at risk family member with the variant) of the p.Arg22Gln variant of KCNH2, an LPV that would cause long QT syndrome, and the p.Arg439Cys variant of LMNA, an LPV that would cause dilated or hypertrophic cardiomyopathy. Possible cosegregation was also detected for the p.Ala688Thr variant of FLNC, a VUS that might cause hypertrophic cardiomyopathy, and for the p.Tyr57His variant of NEBL, a VUS that might cause hypertrophic cardiomyopathy (idiopathic ventricular hypertrophy on autopsy); each of these variants was detected in 2 affected family members of the deceased, but not in only 2 generations.
DISCUSSIONSCD is a leading cause of death in industrialized countries and has an especially profound impact when it occurs in young people. Because of its underlying genetic causes, understanding and preventing SCD requires a multidisciplinary approach.5 More than half of the SCDs in our cohort had a cardiac cause, and cardiomyopathy was the cause of death in more than 20% of the study population. As many as 40% of SCDs of young people remain unexplained (SUDS) despite appropriate histopathological and toxicological analyses, and in this situation it is essential to exclude an underlying heart arrhythmia that might put family members at risk.6,11,12 In our study, 24.4% of SCDs were classified as SUDS, and a genetic variant was found in 70% of affected individuals.
NGS is a very useful tool for the study of SCD, especially for follow-up family screening.13,21 This new tool has replaced traditional molecular autopsy, which examined a handful of genes by Sanger sequencing, most prominently KCNQ1, KCNH2, SCN5A, and RyR214,22. The diagnostic yield of the previous approach was 0-35%.23,24 By comparison, NGS successfully identifies genetic variants in 30% to 35% of individuals dying between the ages of 1 and 35 years and diagnosed with SUDS in the clinical autopsy.15,16,25 NGS offers many advantages over traditional genetic analysis, particularly in comparison with the high probability of false negatives and false positives with exome analysis, which results in high numbers of hard-to-interpret VUS (as many as 13 per deceased individual).
This prospective study demonstrates the diagnostic benefit of comprehensive molecular autopsy of young victims of SCD. As per the study design, deoxyribonucleic acid (DNA) was obtained from all the deceased with an indication for genetic analysis in the histopathological analysis. In contrast with previous practice,25 when blood extraction was not possible or yielded poor quality samples, DNA was obtained from myocardial tissue. By including all consecutive SCD cases in our region and coordinating their study from a single medical examiner's office, the MUSIB program ensures that no cases are missed, again unlike other studies. The NGS analysis used large panels of between 194 and 380 genes related to arrhythmic SCD, a much higher number than used in similar studies. Studies with large NGS panels have been shown to increase the likelihood of finding pathogenic variants by as much as 20%.26,27 The identified genetic variants were classified according to recently published ACMG-AMP criteria.20 If we include VUS as well as P and LP variants, the diagnostic yield of our analysis was 64.5%, and if we include only P and LP variants, the yield was 27.4%. Similarly, in the SUDS subanalysis, the diagnostic yield considering VUS, PV, and LPV was 70%, a figure much higher than reported in other studies, whereas if we consider only P and LP variants the yield drops to 26.6%. Several of the follow-up family screenings demonstrated cosegregation of the identified VUS or LP variants, thus increasing the diagnostic yield to 30.6%. Since family members younger than the deceased are often phenoptypically silent at time of the initial study, the diagnostic yield is sure to increase further as family follow-up continues in future years. It is important to exercise caution when interpreting genetic variants identified in molecular autopsy, and results should always be interpreted within the framework of the clinical family study.27
The incidence of SCD among persons aged ≤ 50 years in the Balearic Islands is 5.8 cases/100 000 individuals/y, which is higher than most reported rates. This is likely in part due to the influence of the large number of tourists who visit the region. However, after excluding deaths of nonresidents, the SCD incidence is 4.2 cases/100 000 individuals/y, which is still higher than that reported in other series; moreover, previous series did not specify whether or not deaths of nonresidents were included.6,11,19 Our analysis did not identify a high-frequency founder mutation that would explain the high SCD incidence. We suspect that the high rate was likely influenced by the fact that more than 50% of autopsies yielded positive toxicology results. Although drug use (mostly cocaine) was the main cause of death in only 10.6% of cases, it may have helped to precipitate death in the context of an underlying condition, whether the toxin was alcohol, an illegal drug, or psychoactive medication.
There is currently no established consensus on the cutoff for defining “young SCD”. Although some studies have used an age threshold of 40 years,28 we set the cutoff at 50 years. This greatly increases the incidence of ischemic heart disease in our cohort, but we wanted to extend the age range to ensure that we detected cases of hereditary SCD that might otherwise have been missed. Indeed, the autopsies of individuals who died between the ages of 40 and 50 years detected 25 potentially hereditary heart conditions: 2 hypertrophic cardiomyopathies, 5 idiopathic ventricular hypertrophies, 1 arrhythmogenic cardiomyopathy, 1 dilated cardiomyopathy, 1 aortic syndrome, and 15 cases of SUDS. Among individuals in this age range and diagnosed with SUDS on autopsy, we identified P or LP variants or VUS in 9 (60%) and P or LP variants in 6 (40%) (3 related to cardiomyopathies, 2 to long QT syndrome, and 1 to catecholaminergic polymorphic ventricular tachycardia). Extending molecular autopsy to this age range thus has the potential to benefit many families, and we therefore believe that this policy should be taken up more generally.
Comparative data from various similar published studies6,11,12,19,25–27,29–34 are summarized in table 2.
Comparative data from published sudden cardiac death series
| ReferenciaStudy | Years | Population | Age range, y | Type of study | Incidence/100 000 individuals/y | N | Cardiac cause, % | Ischemic heart disease, % | Cardiomyopathies, % | SUDS, % | No. genes studied | Sequencing technique | Family screening | Overall diagnostic yield |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lahrouchi et al.29 | 1985-2015 | New Zealand, Denmark, United Kingdom, Netherlands | 1-64 | Retrospective | 302 | 77 | NGS | Yes | 39% | |||||
| Eckart el al.12 | 1998-2008 | United States | ≥ 18 | Retrospective | 902 | 79.3 | 56.6 | 20.7 | No genetic study | No | ||||
| Tester et al.30 | 1998-2010 | United States | 1-69 | Prospective | 173 | 6 | Sanger | No | ||||||
| Winkel et al.19 | 2000-2006 | Denmark | 1-35 | Retrospective | 2.8 | 469 | 67 | 13 | 29 | No genetic study | No | |||
| Margey et al.11 | 2005-2007 | Ireland | 15-35 | Retrospective | 2.85 | 116 | 20.7 | 26.7 | No genetic study | No | ||||
| Jiménez-Jáimez et al.31 | Not specified | Granada (Spain) | Not specified | Retrospective | 30* | 50 | 4-242 | Sanger or NGS | Yes | 33.30% | ||||
| Hertz et al.32 | 2009-2010 | Denmark | 0-50 | Prospective | 15 | 34 | NGS | No | ||||||
| Bagnall et al.6 | 2010-2012 | Australia and New Zealand | 1-35 | Prospective | 1.3 | 490 | 80 | 24 | 16 | 40 | 59-131 | NGS | Yes | 27.00% |
| Sánchez et al.26 | 2012-2016 | Catalonia (Spain) | 0-50 | Prospective | 789 | 52.65 | 55 | NGS | No | 40.40% | ||||
| Hellenthal et al.33 | 2013 | Germany | 1-40 | Prospective | 60 | 20 | 17 | 174 | NGS | Yes | 30% | |||
| Scheiper et al.27 | 2014-2015 | Germany | 1-45 | Retrospective | 99 | 11 | 96 | NGS | No | |||||
| Torkamani et al.34 | 2014-2015 | United States | 0-45 | Prospective | 25 | exome | NGS | Yes | 40% | |||||
| Nunn et al.25 | Not specified | United Kingdom, Spain, Denmark | 1-55 | Retrospective | 59 | 135 | NGS | Yes | 29% | |||||
| Ripoll-Vera et al. (present study) | 2015-2019 | Balearic Islands (Spain) | 0-50 | Prospective | 5.8 | 123 | 52.8 | 38 | 27 | 24.4 | 194-380 | NGS | Yes | 30.60% |
NGS, next generation sequencing; SUDS, sudden unexplained death syndrome.
Studies of SIDS have identified ion channel variants in up to 10% of cases, and more recent NGS analyses indicate the presence of sarcomere variants in 4% to 7% of cases.16,17 In the 5 SIDS deaths in our cohort, we found a genetic variant in 3. All 3 were VUS, 2 related to ion channels and 1 to cardiomyopathy.
In addition to demonstrating the cosegregation of some variants, the family screening detected affected family members and carriers requiring follow-up. To date, an ICD has been placed in 5 patients affected by the same disease as the proband. These devices have been placed as a primary prevention measure in patients with an unfavorable risk profile, and 1 patient has had an appropriate device discharge.
CONCLUSIONSMolecular autopsy and family screening are essential for advancing knowledge of the etiolgy of SCD in young people. This approach allows diagnosis of the underlying genetic cause when this has not been possible with a forensic autopsy or a histopathological analysis without genetic testing, particularly in cases of unexplained SCD. Modern massive sequencing techniques can improve diagnostic yield, but the detection of a VUS in a high proportion of autopsies presents a diagnostic challenge for the future. This challenge can be met by closely monitoring the families of these SCD victims to provide them with better genetic counseling and by using more appropriate risk prevention tools. The establishment of regional multidisciplinary teams is necessary to ensure appropriate histopathological, toxicological, and genetic analysis of young SCD victims and screening of their family members.
- –
SCD in young people is normally caused by genetic heart disease.
- –
Autopsy findings guide the screening of first degree relatives to exclude structural or primary arrhythmogenic cardiomyopathy.
- –
The results of genetic analysis after SCD should be assessed in the context of the activity at the time of death, the personal and family history of the deceased, and the autopsy findings. All of this information is essential for valid interpretation of any genetic variants detected and for establishing the risk status of first degree relatives.
- –
This prospective study of a Spanish series of consecutive cases of SCD in young people is the first of its kind to combine forensic and large-panel NGS molecular autopsy with family cascade screening.
- –
Molecular autopsy gives a high diagnostic yield if it is exhaustive, systematized, and complemented by family screening. This approach diagnoses the cause of SCD in most young victims and can prevent SCD in family members.
- –
The analysis detected genetic variants potentially related to arrhythmogenic cardiomyopathy in 30.6% of cases of SUDS in young people.
Instituto de Investigación Sanitaria de Baleares (IdISBa), Palma de Mallorca, Spain.
CONFLICTS OF INTERESTNone.