
Sudden death is probably the greatest challenge in modern cardiology. After reviewing its history, we describe the epidemiology of sudden death and its associated diseases. We highlight its physiopathologic aspects, including the factors that act on vulnerable myocardium triggering the final arrhythmia, mainly ventricular fibrillation and, to a lesser extent, bradycardia and sudden death. We emphasize the relevance of acute ischemia, ventricular dysfunction and genetic factors, not only in genetic heart disease, but also as triggers of sudden death in acute and chronic ischemic heart disease. Finally, we describe the best way to identify candidates at risk, discuss how to prevent sudden death, and outline the best approach to managing a patient resuscitated from cardiac arrest.
Keywords
Sudden death (SD) is death that occurs unexpectedly within 1 h of the onset of symptoms or when death occurs unwitnessed within 24h of the deceased being seen alive and in a normal state of health before discovery of the body. Some patients die instantly but most have some prodrome.
Historical perspectiveSD has been known for thousands of years.1 In ancient Egypt, more than 4000 years ago, SD was already associated with myocardial ischemia. The Ebers papyrus states: “If a patient has pain in the arm and left side of the chest, there is a threat of death.” In China 2500 years ago, Chio associated SD with an arrhythmia when he said, “An intermittent pulse is a predictor of imminent death.” Around the same time, Hippocrates stated: “Intense chest pain that radiates to the clavicle and back is a sign of poor prognosis.” He was the first writer to introduce the concept of risk factors. He wrote: “Obese individuals are more likely to die suddenly than thin ones.” These concepts have persisted up to the present. We would note in passing that many publications have drawn attention over the centuries to the shock and fear produced by SD.2, 3, 4, 5 In the 14th century, Count Gaston de Foix died suddenly after returning from a hunt during which he had been in contact with icy water. He felt a tightness in the chest and said: “I am a dead man. May God have mercy on me.”5 (Figure 1). In the 18th century, a book written by Lancisi6 on the frequent cases of SD occurring in Rome was published by order of Pope Clement XI. Clinical and autopsy studies showed the association between SD and the presence of chest pain and anatomopathological signs of heart disease. In the late 18th century, Heberden published the first description of “angina pectoris.” In the 19th century, Von Bezold showed that experimental occlusion of the coronary arteries caused cardiac arrest. In the 20th century, Herrick7 described the clinical description of myocardial infarction (MI). Throughout the 20th century, interest in SD increased as its association with coronary disease became clearer,8 as well as the importance of deterioration in ventricular function (heart failure [HF]) in its presentation, regardless of whether this was associated with ischemia or not.

Figure 1. Death of Count Gaston de Foix. 5
Finally, in the second half of the 20th century, it was shown that although ischemic heart disease (IHD) continued to be the cause of SD in at least 80% of cases,9 a group of inherited diseases with structural alterations (cardiomyopathy) or without apparent organic cause (channelopathies) explained many cases of SD in younger people without IHD, whether SD was associated with effort or not.1
EpidemiologySD is probably the greatest challenge in modern cardiology. This is due not only to the immense number of cases (more than 300 000 per year in the United States alone), although its incidence seems to have recently declined and is lower in some Mediterranean countries, such as Spain,10, 11, 12, 13 but also because of its considerable social impact.
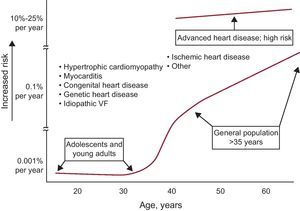
Although SD has been observed even in infants, it is rare in the first decades of life due to its association with impaired repolarization, autonomic nervous system abnormalities, and increased vagal tone. During this period, it usually appears during sports activities14 and in the presence of genetic heart disease (hypertrophic cardiomyopathy, arrhythmogenic right ventricular [RV] dysplasia, arrhythmogenic RV cardiomyopathy, and channelopathies). The incidence of SD gradually increases with age, particularly from 35-40 years onwards, and is particularly high in the acute phase of MI. It is also common in the chronic phase of MI and any heart disease, particularly in the presence of HF (Figure 2).

Figure 2. Association between the incidence of sudden death and age. Note that sudden death is associated with various diseases throughout life. VF, ventricular fibrillation. Reprinted with permission from Myerburg et al. 15
Associated diseasesAs mentioned, acute IHD is often associated with SD in adults. However, in the majority of cases of SD, and outside the setting of acute IHD and inherited heart disease (cardiomyopathies and channelopathies) the presence of HF, or at least ventricular dysfunction, can be demonstrated. HF may be associated with idiopathic cardiomyopathy or found in patients with chronic IHD, hypertension, or other cardiomyopathies, etc. Genetic heart disease can cause SD at any age and, although rare, tends to occur between adolescence and ages 50 to 60 years. However, it should be noted that many cases of SD occur before the age of 35 years (Figure 2). Genetic heart disease manifests more often in men and SD may occur more often during exercise (cardiomyopathies) or while resting or during sleep (channelopathies).
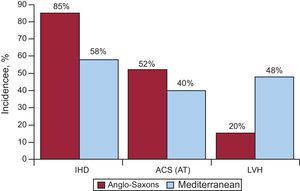
Our group conducted a study within the Instituto de Salud Carlos III group network (the EULALIA study), including 204 cases of SD that occurred in the Mediterranean area (Catalonia and Andalusia).16 This study analyzed the epidemiologic and anatomopathological aspects of diseases associated with SD. Table 1 shows the pathological diagnoses obtained in this group of patients. When compared to a similar study conducted in the United States,17 it is striking that the incidence of IHD detected at autopsy was lower (80%-90% vs 58%); in these cases the incidence of acute thrombosis (anatomopathological basis of acute MI) was also lower (52% vs 40%) (Figure 3). This is in line with current evidence-based knowledge10, 11, 12, 13, 17, 18 that the incidence of IHD in Mediterranean countries is relatively low. This is probably related not only to diet, but also to lifestyle and environmental aspects (Mediterranean culture). In addition, we have noted a greater number of cases of SD involving left ventricular hypertrophy (LVH) (48% vs 20%) in studies conducted in the United States16, 19 (Figure 3). From the clinical point of view, there were fewer cases of angina pectoris in the SD victims in the EULALIA study than in the Maastricht study20 (20% vs 37%), which agrees with the low number of cases of IHD found at autopsy. In our series, the incidence of possible associated genetic diseases was 3% of cases (hypertrophic cardiomyopathy, arrhythmogenic RV), and in about 7% there were no findings at autopsy. This suggests that some of the victims presented a channelopathy.
Table 1. Diseases Associated With Sudden Death: Necropsy Study.
| no., % | |
| Victims of SD | 204 |
| Cardiovascular disease | 183 |
| Heart disease | 161 |
| Coronary heart disease | 119 (58.4) |
| Hypertensive heart disease | 20 (9.9) |
| Valvulopathy | 5 (2.4) |
| Idiopathic LVH | 4 (1.9) |
| Dilated cardiomyopathy | 4 (1.9) |
| Hypertrophic cardiomyopathy | 3 (1.5) |
| Arrhythmogenic RV cardiomyopathy | 3 (1.5) |
| Myocarditis | 1 (0.5) |
| Congenital heart disease | 1 (0.5) |
| Amyloidosis | 1 (0.5) |
| Vascular disease | 22 |
| Pulmonary embolism | 8 (3.9) |
| Aortic dissection | 9 (4.4) |
| Cerebral hemorrhage | 5 (2.4) |
| Noncardiovascular disease | 7 |
| Gastrointestinal disorders | 3 (1.5) |
| Lung disorders | 4 (1.9) |
| No findings | 14 (6.9) |
LVH, left ventricular hypertrophy; RV, right ventricular; SD, sudden death.
Reproduced with permission from Subirana et al. 16

Figure 3. Comparative study of the incidence of ischemic heart disease, acute coronary syndrome with infarction: acute thrombosis and left ventricular hypertrophy in the United States and the Mediterranean area. ACS, acute coronary syndrome; AT, acute thrombosis; IHD, ischemic heart disease; LVH, left ventricular hypertrophy. Reprinted with permission of Subirana et al. 16
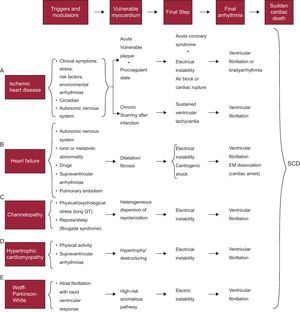
Pathophysiology of sudden deathSD is the final stage of a chain of events leading to cardiac arrest; these are usually ventricular fibrillation (VF) or, less often, severe bradyarrhythmia.21 In all cases, there is a series of modulating or triggering factors that, by acting on vulnerable myocardium, precipitate SD. Figure 4 shows how this chain of events develops in various heart diseases. Briefly, in the presence of vulnerable myocardium, VF is usually precipitated by the action of various modulatory factors and/or triggers, including a specific genetic or environmental context, and/or increased sympathetic activity associated with physical or psychological stress that, although inoffensive under normal circumstances, can precipitate SD in special settings (acute ischemia), hereditary diseases (cardiomyopathies and channelopathies), and so on.

Figure 4. Chain of events precipitating sudden cardiac death and factors associated with various diseases at different stages that lead to sudden cardiac death. AV, atrioventricular; EM, electromechanical; SCD, sudden cardiac death. Adapted with permission from Bayés-Genis et al. 21
Vulnerable Myocardium and Triggering FactorsFigure 4 shows the characteristics of vulnerable myocardium in the heart diseases in which SD more often occurs (IHD, HF, genetic heart disease, and Wolff-Parkinson-White syndrome, although SD is rare in this disease). We now discuss in some detail the 3 factors that are the most common causes of vulnerable myocardium: a) ischemia; b) left ventricular (LV) dysfunction, and c) genetic predisposition. We should bear in mind that the first 2 factors are often interconnected and that the third factor may also play a key role not only in inherited genetic disorders, but also in those that appear to have the most important role, namely ischemia or LV dysfunction.
Ischemic Heart DiseaseAcute ischemia and its effects have a close relationship with the onset of SD, particularly due to the characteristics of the lesions and/or the association with LV dysfunction.
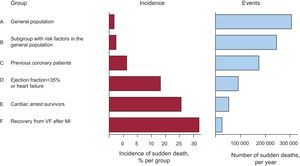
Acute Ischemia and Sudden DeathIn a large number of cases, SD occurs as the first manifestation of an acute coronary syndrome, and these cases form a large percentage of the total burden of SD (Figure 5). In patients with acute MI, the markers of VF are not well defined. We now discuss various studies that have attempted to determine why some patients with acute ischemia, particularly those with ST-segment acute coronary syndrome experience SD but others do not. Naturally, the grade of acute ischemia is of great importance.

Figure 5. The left column shows that the percentage of patients who suffer sudden death is much higher in the high-risk groups (D-F) than in the general population (A, B). However, the total number of cases occurring in the general population (A and B) is greater than in all other groups of patients at risk (C-F), indicating that a large number of cases of sudden death occurs in low-risk groups (A and B). MI, myocardial infarction; VF, ventricular fibrillation.
In the presence of acute ischemia, the severity of ST-segment acute coronary syndromes can be assessed to a great extent by electrocardiogram (ECG).22, 23, 24, 25 ST-segment acute coronary syndromes with grade-3 ischemia22 presents an elevated ST segment with an upwardly deflected S wave. Non-ST-segment acute coronary syndromes with severe ischemia present signs of disease over a circumferential area (≥7 leads with ST depression and ST elevation in VR). This is seen when there is significant left main coronary artery subocclusion or 3 proximal vessels are affected (at least the anterior descending artery and circumflex artery or right coronary artery).24
The grade and duration of ischemia are both relevant. In a study26 that included 21 patients with a total of 120 coronary spasm episodes, we found a direct relationship between ST elevation and the appearance of ventricular extrasystoles. However, due to the briefness of the spasms, although the ST elevation could have progressed to a type of transmembrane action potential, no patient presented ventricular tachycardia (VT) or sustained VF.
The incidence of SD is lower in non-ST-segment acute coronary syndromes than in ST-segment acute coronary syndromes. Patients with non-ST-segment acute coronary syndromes with lower mortality have a low risk score that includes, among other factors, the presence of sinus rhythm with normal heart rate and the absence of ventricular arrhythmias.27
In acute and chronic IHD, the interaction of ischemia with the arrhythmias in the presence of autonomic nervous system abnormalities, LV dysfunction, and genetic and environmental influences28 are relevant factors in the occurrence of VF and SD (Figure 4A). The influence of genetics on the development of IHD and SD is discussed in more detail later in the article.
Acute coronary syndromes often progress to MI. Current treatments have reduced the number of patients with severe long-term complications of infarction, such as HF and depressed ejection fraction (EF). However, IHD is still frequently associated with cardiac SD, although it is unclear whether a number of factors enhance this association or an acute ischemic episode is the direct cause of SD. This is probably the most important challenge in modern cardiology.
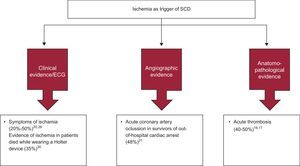
In the United States, between 80% and 90% of cases of SD are associated with IHD, but this association is lower in the Mediterranean area.16 However, global clinical data (presence of angina),20, 29 electrocardiographic data (ST changes on Holter monitoring)30 and angiographic data (acute thrombosis)31 show that acute ischemia is the direct causal factor of SD in less than 50% of cases (Figure 6). Moreover, in about 50% of cases of SD there is also anatomopathological evidence of acute infarction (fresh thrombus).16, 17. These findings are important because they show that only about 50% of SD cases are due to an acute ischemic episode. In most other cases of SD in patients with IHD, the cause of death is probably sustained VT due to reentry around an old infarction lesion, triggering myocardial VT or VF.

Figure 6. Relevance of acute ischemia as a trigger of sudden death: clinical, electrocardiographic, angiographic, and anatomopathological tests. ECG, electrocardiogram; SCD, sudden cardiac death.
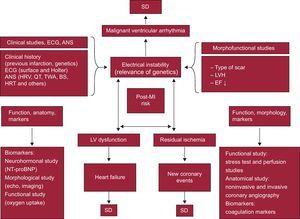
Sudden Death in Patients After InfarctionThe risk of severe ventricular arrhythmias and SD is particularly high in the first 6 months post-acute MI. Although risk stratification in chronic IHD patients, particularly for SD,32 is continually evolving, in post-MI patients there are 3 three major risk factors: a) the presence of residual ischemia (risk of new ischemic event;33 b) poor ventricular function (risk of HF),34 and c) electrical instability (risk of severe arrhythmias and SD).34, 35
Figure 7 shows the factors that can be risk markers of these complications, as well as the techniques used to study them.1, 24 According to the triangle shown in this figure, the following measures are the most important in risk stratification:
• Measures of electrical instability: a) morphological and functional studies (type of scar, presence of LVH and depressed LVEF, infarct size, etc.); b) clinical aspects (age, family history, previous MI and surface ECG data, such as the presence of Q wave and QRS width, etc.); c) Holter monitoring (presence of ventricular extrasystoles and heart rate [HR], c.), and d) studies of autonomic nervous system disorders, such as HR variability and baroreflex sensitivity, which indicate decreased tone and vagal reflexes,36 decreased vagal response,37 and increased sympathetic tone. Other factors studied include T-wave alternans38, 39 and HR turbulence.40
• Measures of LV function, obtained from: a) LV function imaging techniques, such as echocardiography; b) LV functional capacity via exercise testing (oxygen consumption), and c) the measurement of neurohormonal markers, such as amino-terminal pro-B-type natriuretic peptide (NT-proBNP).
• Measures of residual ischemia: a) anatomical studies (noninvasive and invasive coronary angiography); b) functional studies (ECG exercise testing and perfusion imaging techniques), and c) coagulation biomarker tests. The presence of silent ischemia may also be a marker of SD over the long term (5-year risk).41

Figure 7. Risk stratification in patients after a heart attack, according to the presence of the following factors and their interactions: a) electrical instability; b) left ventricular dysfunction, and c) residual ischemia. The various factors that precipitate complications and sudden death and the tests used in their detection are shown. ANS, autonomic nervous system; BS, baroreflex sensitivity; ECG, electrocardiogram; EF, ejection fraction; HRT, heart rate turbulence; HRV, heart rate variability; LV, left ventricle; LVH, left ventricular hypertrophy; MI, myocardial infarction; NT-proBNP, amino-terminal pro-B type natriuretic peptide; SD, sudden death; TWA, T-wave alternans.
However, it is noteworthy that even in the presence of some of these classic triggers—such as the number of premature ventricular complexes (PVC), equal infarct size, and other clinical characteristics—some patients present nonsustained VT episodes, sometimes frequently, whereas other patients do not. It is clear that environmental and genetic aspects and autonomic nervous system abnormalities, sometimes very subtle, can have an influence, although the measurement of these factors has a low positive predictive value. However, it has recently been shown that there are electrophysiologic and anatomic differences in the lesion substrate that may explain the tendency to experience VT. It has been shown42 that, given the same environment-related PVC and similar clinical features, patients who have experienced infarctions involving more scar tissue, more fibrosis, and more fractionated electrograms present sustained VT more frequently. This paper also demonstrates that the anatomical substrate (scar features) is more important than the presence of environment-related PVC (triggering and modulating factors) in inducing sustained VT. This study highlights the need for noninvasive markers to identify the presence of this substrate. Some have already been mentioned, such as the presence of a fragmented QRS and intraventricular block. This suggests that extensive ablation, including areas with myocardial bridges between the lesion areas, is probably the most valid and effective approach in the case of repetitive VT.
It is also striking37 that in patients with previous infarction but without residual ischemia, the incidence of severe ventricular arrhythmias was much higher in inferior infarcts than in anterior infarctions. This higher incidence of ventricular arrhythmias in inferior MI is even more unexpected if we take into account that LVEF was significantly higher in inferior than in anterior MI. One possible explanation is that in the lower wall there is a high density of receptors with vagal activity that are cardioprotective. Thus, after an infarction in this area, the protective effect of the vagal response is lower, which may explain why the risk of ventricular arrhythmias is greater.
Ventricular Dysfunction and Heart FailureExcept for cases of SD in patients with genetic heart disease, and those associated with acute MI, particularly with ST elevation, most patients who die suddenly, regardless of etiology, have impaired LV function, often with HF and in general with depressed EF (systolic HF).
Figure 4B shows the chain of events triggering ventricular arrhythmias (VT/VF) which cause SD in patients with HF (New York Heart Association class II-III).21
Approximately 40% of cardiovascular deaths that occur in patients with HF are sudden. The other cardiovascular deaths are due to HF progression. Between 20% and 30% of patients with New York Heart Association grade II-III HF die within 3 years of follow-up, and about half of the cases are due to SD (hearts still in a good state of health). SD in patients in functional class II and III is due in particular to ventricular arrhythmia (VT/VF) as the final arrhythmia, but SD in patients in functional class IV is explained by bradyarrhythmias. This may explain the ineffectiveness of antiarrhythmic drugs to prevent SD in patients with HF and New York Heart Association functional class IV.
Although there are conflicting results, most current studies show that SD is more often observed in patients with systolic HF, especially in patients with ischemic dilated cardiomyopathy (DC), than in patients with diastolic HF.
Moreover, there is much evidence on the role of different autonomous nervous system abnormalities (HR variability, HR turbulence, sinus tachycardia) as risk markers of SD,43 although their positive predictive value is low. There is also evidence that microvolt T-wave alternans may be useful as a risk marker of SD.38, 39
There is a correlation between increased sympathetic innervation, which can be detected by 123I-labeled m-iodobenzylguanidine scintigraphy, and improvement in HF prognosis and decreased ventricular arrhythmias.
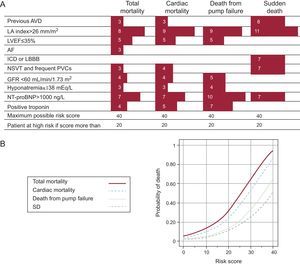
Recently, we published (Instituto Carlos III group network) a scale of the risk of death in patients with HF (MUSIC study).44 This study was designed to analyze the clinical, electrocardiographic (12-lead ECG and Holter monitoring), echocardiographic and biochemical variables implicated in the onset of SD in patients with HF. This was a prospective study of 992 patients with systolic HF (75%) or diastolic HF (25%). In total, 78% of patients were in New York Heart Association functional class II at the time of inclusion. Patients were followed up for 44 months and total death, cardiovascular death, death from pump failure, and SD were analyzed. Among the variables analyzed, 10 had independent prognostic significance (a different numerical value for each variable). Note that only atrial dilatation and NT-proBNP formed part of the score for total death. The combination of these variables allowed us to establish a risk score, as shown in Figure 8. Using this risk score, 2 well-defined populations were identified, with scores<20 and >20; these populations were considered to be at low risk and high risk, respectively. Recently, a new serum marker of SD, ST2,45 has been identified, which should further improve risk stratification of SD. However, further studies are needed in this field to validate these results. The potential economic impact of decreasing the number of implantable cardioverter defibrillator (ICD) procedures would also warrant more studies of this kind in the future.

Figure 8. A, risk score for predicting different types of death. The left column shows the factors used to determine the score. B, mortality curves for different types of death over a 3-year period. Reprinted with permission from Vasquez et al. 43 AF, atrial fibrillation; AVD, arterial vascular disease; GFR, glomerular filtration rate; ICD, intraventricular conduction defect; LA, left atrium; LBBB, left bundle branch block; LVEF, left ventricular ejection fraction; NSVT, nonsustained ventricular tachycardia; PVC, premature ventricular complexes; SD, sudden death.
GeneticsWe have already referred to possible genetic interactions in the development of acute and chronic IHD and SD. We believe that one of the keys to future SD research is to study the influence of genetics in SD patients with ischemia and/or HF.
The potential role of genetic factors in the development of VF during non-ST-segment acute coronary syndromes has recently been described.46 An association has been postulated between the density of the lto current, responsible for Brugada syndrome and other J-wave syndromes,47 and the risk of VF during ST-segment acute coronary syndromes. This could explain both the high incidence of VF among men with inferior infarction with RV involvement. The Ito current is far more prominent in men, and more prominent in the RV than in the LV. We believe that these genetic interactions probably play a key role in explaining the occurrence of fatal ventricular arrhythmias. In fact, it has been proposed that the fundamental mechanisms underlying ST elevation and the initiation of VF are probably similar in the initial phase of ST-segment acute coronary syndromes and the J wave syndromes that have a genetic substrate. In both cases, the heterogeneous dispersion of repolarization and phase 2 reentry are the triggers and reentrant substrate underlying the onset of VT/VF.48
Three major studies have identified genetic variants associated with the risk of SD in patients with IHD. The first study, conducted in the Netherlands,49 identified a locus on chromosome 21, in which the rare allele of SNP rs2824292 was associated with an increased risk of VF in patients with acute MI (odds ratio=1.49; 95% confidence interval, 1.14-1.95). The closest gene to this locus is CXADR, which encodes a viral receptor previously implicated in myocarditis and DC and that modulates cardiac electrical impulse conduction. The other 2 studies, conducted in populations in the United States,50, 51 found that the rare allele of SNP rs3864180 located in the GPC5 gene is associated with a decreased risk of SD in patients with IHD (hazard ratio=0.85; 95% confidence interval, 0.74-0.98). The presence of this allele was also associated with a shorter duration of the QT interval. Finally, the rare allele of SNP rs4665058 is associated with an increased risk of SD in patients with chronic IHD (odds ratio=1.91).
We now discuss SD occurring in genetically induced diseases. These include52 cardiomyopathy due to cardiac protein abnormalities, such as hypertrophic cardiomyopathy, arrhythmogenic RV dysplasia, noncompaction cardiomyopathy and certain forms of DC; specific conduction system disease (Lev-Lenègre) and channelopathies (disorders without overt structural disease or with minimal structural disease) due to isolated ion channel abnormalities, such as long and short QT syndromes, Brugada syndrome and catecholaminergic VT, and probably familial atrial fibrillation; and other types of SD due to malignant ventricular arrhythmias, such as familial torsade de pointes VT and idiopathic VF, and even some cases of sinus bradycardia of genetic origin and possibly Wolff-Parkinson-White syndrome.
Some of the better-known channelopathies show a characteristic phenotypic pattern on ECG, which often leads to the correct diagnosis and even to identification of the gene involved. However, it has been reported that some of the so-called channelopathies may present some ultrastructural changes. Histological abnormalities have recently been described53 and subtle abnormalities in RV morphology detected on cardiac magnetic resonance imaging54 in asymptomatic relatives of patients with Brugada syndrome who carry mutations on the SCN5A gene. In addition, Papavassiliu et al.55 have demonstrated the existence of certain ultrastructural abnormalities in Brugada syndrome. This confirms that the phenotypic manifestation of Brugada syndrome and its pathophysiology is more complex than initially thought and that the apparently isolated ionic abnormalities may be accompanied by ultrastructural changes detected on magnetic resonance imaging.
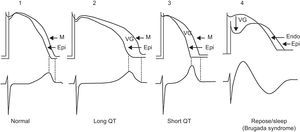
Channelopathies may incur the risk of triggering different types of VT/VF and SD. In the long QT syndrome, short QT syndrome, and Brugada syndrome, VT/VF is explained by the heterogeneous dispersion of repolarization and occurs in some areas of the myocardium more than in others, regardless of whether the dispersion is transregional56 or occurs in the thicker part of the LV wall (transmural dispersion).57 This heterogeneous dispersion of repolarization increases the possibility of a premature beat, which may induce VT/VF, most probably via a phase 2 reentry event (Figure 9). It has been shown that heterogeneous dispersion of ventricular repolarization explains the increase in the Tpeak-Tfinal interval and that this parameter is a marker of poor prognosis and ventricular arrhythmias.

Figure 9. Example of phase 2-3 reentry when reentry occurs due to the heterogeneous dispersion of repolarization. According to Antzelevitch, 57 dispersion occurs at the transmural level and when the transmembrane action potential of the M cells is longer than that of the rest of the wall. This heterogeneous dispersion of repolarization accounts for the appearance of a voltage gradient between the area with a more prolonged transmembrane action potential and the area where transmembrane action potential is shorter (the epicardium), and for the possibility of ventricular tachycardia/ventricular fibrillation occurring in patients with long QT syndrome and short QT syndrome. In Brugada syndrome, the heterogeneous dispersion of repolarization occurs between the endocardium and epicardium of the right ventricle at the start of phase 2, creating a voltage gradient due to the predominance of the transient outward current Ito. Endo, endocardium; Epi, epicardium; M, M-cell layer; VG, ventricular gradient.
Hypertrophic cardiomyopathy is usually a family disease of genetic origin, characterized by cardiac myocyte proteins abnormalities, leading to myocardial disarray, cardiac hypertrophy, and an increased incidence of SD.58
DC can be idiopathic (30%-40% of cases) or may be due to a range of causes (for example, infections, toxins, drugs, autoimmune processes, or neuromuscular, metabolic, and mitochondrial diseases), but the most common causes are chronic IHD and/or hypertension. However, the new classification of cardiomyopathies has excluded58 (in the absence of convincing arguments, in our opinion) myocardial disease as a direct result of other cardiovascular diseases such as hypertension, IHD, valvular heart disease, and congenital heart disease.
Evidence exists of a familial link or a shared environment in up to 35% of idiopathic DCs.59 More than 20 mutations have been identified in different genes, among them the lamin A/C gene, which is present in 30% of patients with DC and is associated with a high incidence of SD. In addition, genetic associations have been reported between DC and Duchenne muscular dystrophy, Emery-Dreifus muscular dystrophy, and Becker muscular dystrophy, all of which can cause cardiac abnormalities that lead to DC and IHD.60
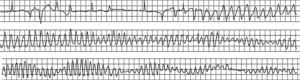
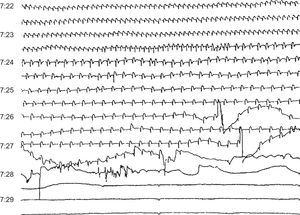
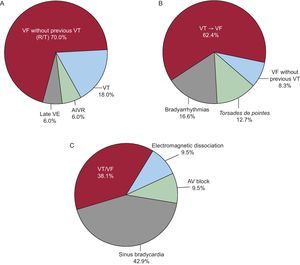
Final ArrhythmiasThe final arrhythmias that trigger SD are not always the same (Figure 10, Figure 11, Figure 12, Figure 13) and are not always acute in different clinical settings. We conducted a study in which we reviewed the tapes of 157 ambulatory patients who died from SD while wearing Holter monitors30 (Figure 14B and Table 2), finding that almost two thirds of these cases were due to sustained VT that precipitated a VF, generally in the presence of a rapid baseline HR (sinus tachycardia or rapid atrial fibrillation), indicating an increase in sympathetic activity (Figure 10). In contrast, VF without a precipitating VT, which is often associated with acute IHD, commonly presents as a result of PVC with an R-on-T phenomenon (Figure 11). We observed this pattern in less than 10% of the ambulatory patients. Interestingly, in 13% of patients, SD was due to a torsade de pointes VT, usually in patients without severe heart disease but who were taking class I antiarrhythmic drugs for rare ventricular arrhythmias, often isolated PVC (proarrhythmic effect) (Figure 12). We believe that if the study were repeated today the number of such cases would be much lower, as evidence from the CAST study63 showed that type I antiarrhythmic drugs are dangerous, especially in cardiac patients. For this reason, the use of class I antiarrhythmic drugs in patients after acute MI is currently far less common. Finally, when severe bradyarrhythmia was the cause of SD (15% in our study) (Figure 13), this was more often due to progressive depressed sinus node automaticity and atrioventricular junction automaticity than to atrioventricular block.

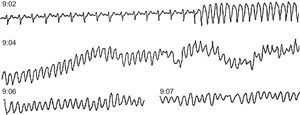
Figure 10. Ambulatory sudden death due to ventricular fibrillation in a patient with coronary heart disease treated with amiodarone for frequent premature ventricular contractions. At 9:02 the patient suffered sustained monomorphic ventricular tachycardia, followed by ventricular fibrillation at 9:04 after an increase in the rate of ventricular tachycardia and the width of the QRS complex.

Figure 11. Ambulatory sudden death due to primary ventricular fibrillation caused by a ventricular extrasystole with a short coupling interval, after a postextrasystolic pause (1120ms) longer than the previous one that had caused an isolated premature ventricular beat. Note that the sequence of events began with an atrial extrasystole that led to the first shorter pause (860ms).

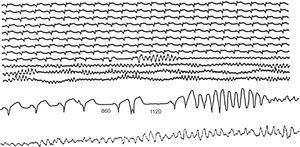
Figure 12. Initiation of torsade de pointes ventricular tachycardia in a woman treated with quinidine for bursts of nonsustained ventricular tachycardia without coronary disease. In this patient, torsade de pointes ventricular tachycardia triggered ventricular fibrillation.

Figure 13. Sudden death due to progressive bradycardia in a patient with acute infarction and probable electromechanical dissociation.

Figure 14. Sudden death: final arrhythmias. A, in patients with acute ischemic heart disease (Adgey et al. 61 ); B, in outpatients, in 80% of patients with depressed ejection fraction (Bayés de Luna et al. 30 ); C, in patients with advanced heart failure (Luu et al. 62 ). AIVR, accelerated idioventricular rhythm; AV, atrioventricular; VE, ventricular extrasystoles; VF, ventricular fibrillation; VT, ventricular tachycardia. Reproduced with permission from Bayés de Luna. 52
Table 2. Clinical Characteristics of Patients Wearing Holter Monitors Who Died Suddenly.
| Group I, primary VF and VT→VF (n=112) | Group II, torsade de pointes VT (n=23) | Group III, bradyarrhythmias (n=26) | |
| Age, years | 58 | 60 | 70 |
| Men | 76% | 40% | 39% |
| Coronary patients | 90% | 50% | 70% |
| No overt heart disease | 4% | 22% | 10% |
| Antiarrhythmic therapy | 51% | 70% | — |
VF, ventricular fibrillation; VT, ventricular tachycardia.
Adapted from Bayés de Luna et al. 30
Figure 14 shows the final arrhythmias that caused SD in patients with different clinical characteristics: patients in a mobile coronary care unit with symptoms of acute ischemia outside hospital61 (Figure 14A); ambulatory patients (Holter monitoring)30 (Figure 14B); and patients admitted with severe HF (Figure 14C).62 In the first case (A), the number of patients with VF without prior VT was much larger than in the ambulatory group in our study (B), most likely due to the fact that all the patients in group A were in the acute phase of MI. In patients with advanced HF (group C), there were many more cases of SD due to severe bradyarrhythmia. This could explain the ineffectiveness of antiarrhythmic drugs to prevent SD in patients with advanced HF. In our series, 80% of patients had depressed EF, but in general their functional capacity was acceptable (Figure 14B). These patients were “too healthy to die” and many cases of SD could have been prevented with proper treatment, which sometimes means not prescribing class I antiarrhythmic agents. We should recall the Hippocratic dictum: “Primum non nocere”.
Our results were similar to those found in patients with ICD whether or not they were receiving cardiac resynchronization therapy. In this case, rapid VT also frequently occurred and was treated with antitachycardia pacing.64, 65 On the other hand, in a small series of post-acute MI patients with event monitors (implantable loop recorders) with EF<40% who died suddenly, in most cases SD was due to primary VF not preceded by VT.66 However, no information is available on the clinical events around the time of death. In this series, the limited amount of information and small number of cases hinders comparison with our results. On the other hand, in most of the patients who died from different types of bradyarrhythmias, death occurred more than 1 h after the onset of symptoms.15
How to identify patients at riskWe know much more about identifying and preventing SD in patients at risk (history of cardiac arrest, genetic heart disease, some patients after acute MI, patients with HF, etc.), than we do about how to predict cases in which SD appears as the first manifestation in the general population.67 Figure 5 shows that these cases (A and B) represent more than 50% of patients with SD. Many of these patients present acute MI whose initial manifestation is SD.
Thus, it is extremely difficult to identify candidates at risk of SD among individuals who have not previously had symptoms or who do not have serious risk factors, since it is not possible to screen the entire population. Currently, the most we can aspire to is the following: a) conduct a detailed study of the relatives of patients who have had SD; b) when seeing a patient for another reason, obtain the complete medical history to ascertain if there is a family history of genetic heart disease, IHD, or obvious risk factors, and c) as part of any first medical appointment with an adult patient, conduct a thorough physical examination and order any relevant laboratory tests (blood tests to identify any risk factors, especially elevated cholesterol, blood sugar, and blood pressure, and an ECG).
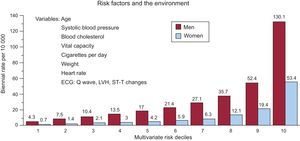
Discovering candidates at risk of SD in the general population is one of the greatest challenges of modern cardiology. The Framingham study, conducted in the general population, established the following68: a) the presence of ECG abnormalities, especially intraventricular block and LVH, substantially increases the risk of SD, primarily in men, and b) a multivariate analysis of risk showed that the risk of SD increased according to the number of risk variables presented, especially in men (Figure 15). It has recently been reported that marked elevations of NT-proBNP are more indicative of a high risk of SD in women than some of the classic risk factors (lipid abnormalities).69

Figure 15. Risk of sudden death in the Framingham study according to multivariate risk in men and women. ECG, electrocardiogram; LVH, left ventricular hypertrophy. Adapted with permission from Kannel et al. 68
Preventing sudden deathIt should be obvious that identifying candidates at risk of SD is the best way to prevent it. Various groups are at risk of SD, and within these groups it is crucial to select those who are most at risk in order to consider the option of implanting an ICD once all the pharmacologic or other strategies have been exhausted, such as ablation of an anomalous pathway or arrhythmogenic focus (eg, VT). Although an ICD does not prevent the occurrence of fatal arrhythmia, if one occurs then it may be possible to avoid SD.
All the factors involved in the prevention of SD in patients at high risk are well known (Figure 5, Figure 7). However, it is far more difficult to prevent SD in the general population.70, 71, 72, 73 This requires effectively fighting the most common diseases associated with it, such as IHD, HF, and genetic heart disease. The prevention of IHD should begin in childhood via health education programs that promote a heart-healthy lifestyle by encouraging physical exercise and proper diet in order to help fight obesity and prevent the occurrence of risk factors. It is also important to effectively combat elevated cholesterol levels, hypertension, and diabetes mellitus, and to prevent HF or provide effective treatment from the moment it begins. In addition, we need to know the key factors that allow us to diagnose the genetic heart diseases implicated in SD; this includes increasing awareness of the relevance of a personal or family medical history of syncope or SD and the fact that a simple ECG pattern may suggest that our patient is a possible candidate for SD.
In fact, if we speculate about the future, what would be of great help in reducing SD would be an atheromatous plaque stabilizer, perhaps a chemical additive in food or water or which could even be widely administered to the whole population, which would stabilize the fibrous cap of active atherosclerotic plaque to reduce the likelihood of erosion or rupture and SD.74
Patient care after resuscitation from cardiac arrestPatients who are resuscitated after cardiac arrest outside hospital should be sent to a tertiary referral hospital for comprehensive studies to identify the causes by exhaustive clinical studies, including invasive and noninvasive tests. This approach is appropriate for patients who have suffered cardiac arrest with no overt underlying heart disease, although systematic genetic studies are not recommended in these patients. If they are available and economically feasible, their use is recommended in the relatives of patients with genetic heart disease.
In most cases, the cause of the cardiac arrest is VF. Therefore, we should prevent the first episode and coordinate the prevention of future episodes. Specific decisions are sometimes required. For example, if VF appears to be associated with ischemia, revascularization should be considered. Other mechanisms that may trigger VF should also be ruled out; for example, in the case of rapid atrial fibrillation in patients with Wolff-Parkinson-White syndrome, ablation of the accessory pathway is obligatory. If the patient has suffered syncope, a complete examination is needed.75 Naturally, in patients in whom cardiac arrest is due to a passive arrhythmia, the patient should undergo immediate pacemaker implantation.52
In all other cases, if it is confirmed that cardiac arrest is due to VT/VF without obvious cause, ICD implantation is recommended and, if needed, resynchronization therapy administered. We recommend consulting the current guidelines of the American76 and European77 scientific societies on ICD implantation.
Conflicts of interestNone declared.
Received 30 March 2012
Accepted 31 March 2012
Corresponding author: Institut Català de Ciències Cardiovasculars, Avda. S. Antoni M. Claret 167, 08025 Barcelona, Spain. abayes@csic-iccc.org