Massive DNA sequencing, also known as next-generation sequencing, has revolutionized genetic diagnosis. This technology has reduced the effort and cost needed to analyze several genes simultaneously and has made genetic evaluation available to a larger number of patients. In hypertrophic cardiomyopathy, genetic analysis has increased from the 3 main genes implicated in the disease (MYH7, MYBPC3, TNNT2) to sequencing of more than 20 related genes. Despite the advantages of acquiring this additional information, many patients show variants of uncertain significance (mainly amino acid changes), which may also be present in at least 1 healthy control undergoing genome sequencing. This will be a dead-end situation unless the variant can be demonstrated to be associated with the disease in the patient's family. In the absence of clear evidence that these variants are truly pathogenic, they cannot be used for reliable genetic counselling in family members. Massive sequencing also enables identification of new candidate genes, but again, the problem of variants of uncertain significance limits the success of these assessments.
Keywords
Hypertrophic cardiomyopathy (HCM) is the primary form of left ventricular hypertrophy.1–3 In HCM, hypertrophy develops when the cardiac muscle is “pressured” to increase its contractile activity to compensate for a reduction in this capacity due to endogenous reasons.4 The most common of these is abnormal isoforms of the contractile proteins, but the origin of HCM can also be related to changes in the mechanisms of energy production and regulation of myocardial contraction.5 As HCM is an essential disease, mutations will obviously be found in genes encoding myocardial contractile proteins.6 Hypertrophic cardiomyopathy is a common cause of sudden cardiac death in young adults; hence, there is considerable general interest in characterizing the genetic basis of this disease.7
Since the discovery that a large percentage of patients with HCM show MYH7 and MYBPC3 mutations, research has identified additional, less common gene mutations that have been incorporated in the diagnostic arsenal of this condition.7–12 On the practical side, there has been a technological revolution with the development of new techniques for DNA sequencing. These methods, known as massive parallel sequencing or next-generation sequencing, enable analysis of several genes simultaneously and at a lower cost than other available techniques.13–16 Nonetheless, despite the evident advantage of having access to more extensive information about genetic variation, massive sequencing may not always resolve the question of whether a patient has a mutation that would explain the presence of the disease.
The aim of this review was to provide an overview of the strengths and weaknesses of massive sequencing in the genetic diagnosis of HCM.
WHAT DO WE KNOW ABOUT THE GENETICS OF HYPERTROPHIC CARDIOMYOPATHY?Before the development of massive sequencing technology, HCM mutations were detected by amplifying multiple fragments of each potentially related gene and reading the fragments separately using Sanger sequencing.17,18 Because this requires enormous technical and economic resources,, most laboratories to limit the analysis to sequencing the most frequently mutated genes (MYH7, MYBPC3, and TNNT2), and when these test negative, others can be evaluated.11,12 Studies using this method have yielded the following conclusions:
- 1.
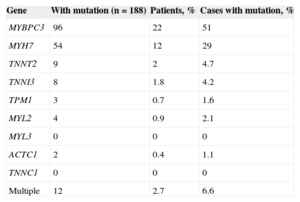
Approximately 40% to 60% of HCM patients show mutations in 1 or more of the genes known to be associated with the disease. The MYH7 and MYBPC3 genes are most often affected, and mutations in these genes account for around 50% of all mutations identified in HCM patients (Table 1).
Table 1.Results of the Analysis of the Main Sarcomeric Genes in 444 Unrelated Patients Evaluated in Hospital Universitario Central de Asturias (Spain)
Gene With mutation (n=188) Patients, % Cases with mutation, % MYBPC3 96 22 51 MYH7 54 12 29 TNNT2 9 2 4.7 TNNI3 8 1.8 4.2 TPM1 3 0.7 1.6 MYL2 4 0.9 2.1 MYL3 0 0 0 ACTC1 2 0.4 1.1 TNNC1 0 0 0 Multiple 12 2.7 6.6 - 2.
Mutations are more likely to be detected in patients with a related family history and severe early-onset HCM, whereas they are less apt to be found in sporadic, milder cases developing at a more advanced age. Furthermore, patients with sporadic disease are more likely to show variants whose pathogenic role is difficult to confirm, as there are no affected family members to provide evidence of transmission (among other reasons). Such variants, which have been categorized as gene variants of uncertain significance (VUS), pose a problem for genetic counselling because it is impossible to conclusively establish that they are the cause of the disease in the index case (Figure 1).
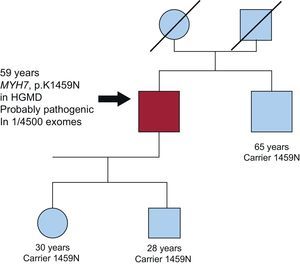
 was a man diagnosed with myocardial hypertrophy of 19mm at the age of 69 years. Sequencing of the 9 sarcomeric genes yielded a p.K1459N change in MYH7. This change has been found in other patients with MCH, and was classified as probably pathogenic by the prediction software. It is present in 1 of the sequenced exomes. One of the patient") Figure 1.
Figure 1.Family with carriers of an MYH7 variant of uncertain significance. The index case (arrow) was a man diagnosed with myocardial hypertrophy of 19mm at the age of 69 years. Sequencing of the 9 sarcomeric genes yielded a p.K1459N change in MYH7. This change has been found in other patients with MCH, and was classified as probably pathogenic by the prediction software. It is present in 1 of the sequenced exomes. One of the patient's brothers, who also harbored the variant, showed no hypertrophy and remained asymptomatic at the age of 65 years. Based on this accumulated information, it cannot be concluded that the mutation is linked with hypertrophic cardiomyopathy and is not a rare polymorphism (nonpathogenic). HGMD, Human Gene Mutation Database.
(0.09MB). - 3.
Contrary to what was indicated in earlier studies, there will be no clear relationship between the mutated gene and the severity of HCM. In general, it is better to consider the degree of severity of a particular gene.19 However, to establish a relationship between a mutation (genotype) and the phenotype, a sufficient number of carriers is required, and this is only possible with a few mutations. Among these are the 25-nucleotide deletions in MYBPC3, p.R453C and p.G716R in MYH7, and p.R92W inTNNT2, all of which are clearly pathogenic and associated with a poor prognosis.7,20–23 These mutations are distributed throughout the world, but others are exclusively found in specific geographic regions and they may also be present in a large enough number of patients to enable an analysis of their link with the phenotype. For example, in Asturias (Spain), 8% of (unrelated) HCM patients are carriers of a stop codon in MYBPC3 (p.G263X), which is associated with a generally benign phenotype.24 In Iceland, 58% of patients are carriers of the c.927-2A>G MYBPC3 mutation, related to a heterogeneous phenotype.25 A mutation in the first base of intron 23 of MYBPC3 is common in Spanish patients and is characterized by HCM onset in middle age and a poor prognosis, with a high risk of sudden cardiac death.26
- 4.
Many variants classified as possible mutations are exclusive to single patients and their families (private mutations). In these cases it may be impossible to establish a genotype-phenotype relationship, which is one reason why clinical guidelines advise against using them for risk stratification and therapeutic decision-making in patients harboring them.3,8,27,28 One obstacle for interpreting the genetic findings in these patients is that there is no dedicated HCM database where all the known mutations are recorded, and access to such information is only possible through publications or subscription to the HGMD (Human Gene Mutation Database). Therefore, a laboratory might classify several variants as new findings, when actually they had been found (but not reported) in other patients.
- 5.
Up to 5% of HCM patients may harbor 2 or more mutations. In our series of 444 patients, 12 (3%) were double carriers of MYH7, MYBPC3 or TNNT2.12 Other authors have described a similar percentage (2% in the Mayo Clinic series).11 The presence of 2 or more mutations has been related to greater disease severity.29,30 As clinical guidelines consider detection of more than 1 mutation indicative of a poor prognosis, it is admissible to use this genetic information to guide therapeutic decision-making.3,8
 was a man diagnosed with myocardial hypertrophy of 19mm at the age of 69 years. Sequencing of the 9 sarcomeric genes yielded a p.K1459N change in MYH7. This change has been found in other patients with MCH, and was classified as probably pathogenic by the prediction software. It is present in 1 of the sequenced exomes. One of the patient")
In certain situations, the presence of more than 1 mutation should be evaluated in a family even though only a single mutation is found in the index case (Figure 2). When relatives not carrying a mutation are seen to be affected by the disease, a second mutation should be suspected. Furthermore, the presence of 2 mutations would explain heterogeneity in the phenotypic manifestations in the same family, in which carriers of a single mutation would manifest a less severe phenotype than double carriers. To increase the yield of genetic study in a family, it is advisable to seek mutations in the member with more severe HCM developing at an earlier age.
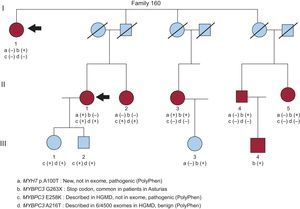
 was a woman harboring the p.G263X mutation in MYBPC3 (variant b). However, an affected niece (II.1) was not a carrier of the mutation, and the 9 genes were sequenced in her samples. Three variants were found, with MYH7 p.A100T (a) and MYBPC3 p.E258K (c) probably being pathogenic. HGMD, Human Gene Mutation Database.")
Family with several possibly pathogenic changes. The index case (I.1) was a woman harboring the p.G263X mutation in MYBPC3 (variant b). However, an affected niece (II.1) was not a carrier of the mutation, and the 9 genes were sequenced in her samples. Three variants were found, with MYH7 p.A100T (a) and MYBPC3 p.E258K (c) probably being pathogenic. HGMD, Human Gene Mutation Database.
One particularly intriguing possibility is that many patients with apparently sporadic forms of HCM may actually be carriers of 2 or more mutations having low penetrance. That is, variants that have a weaker pathogenic effect and predispose to less severe forms of the disease when they occur alone, but that show greater pathogenicity when combined. Individuals with a single mutation may remain asymptomatic to an advanced age.
MUTATIONS THAT SPEAK FOR THEMSELVESWhen initiating genetic study in a patient with HCM, one can predict the probability (high or low) of finding some type of mutation; that is, clear information regarding the genetic cause of the disease. One only has to determine whether there are various relatives with confirmed HCM in the family or, conversely, that the case is sporadic. If, in addition to a family history, the patient has a severe phenotype and early onset of the disease, there is a high probability of finding a genetic change classified as a mutation. It is such families with several affected members that have enabled the identification of the sarcomeric genes linked to HCM.31–34
In patients not meeting these conditions, it is very likely that mutations will not be found or that some VUS will be detected.11,12 There is little uncertainty regarding the pathogenicity of a DNA base change that results in an amino acid being replaced by a stop codon, or a change in the first or last base of an intron (splice site mutation, affecting messenger ribonucleic acid processing and giving rise to an anomalous protein). In HCM, most nucleotide changes lead to a substitution of one amino acid for another (missense variants), as is the case of those mentioned above: p.R453C and p.G716R in MYH7 and p.R92W inTNNT2. Substitutions of this type are undoubtedly pathogenic, among other reasons because they are detected in HCM-affected patients and families, but not in healthy individuals. Most of these changes have been seen in families with several affected members, and once they have been identified, genetic counseling does not pose a problem in carriers (predisposed to having the disease) and noncarriers (having the same risk of developing HCM as the general population).35 It might even be possible to provide a preimplantation diagnosis for the offspring of these carriers.36 One could say that these DNA changes constituting true mutations “speak for themselves” because of their familial and phenotypic context (Figure 3).
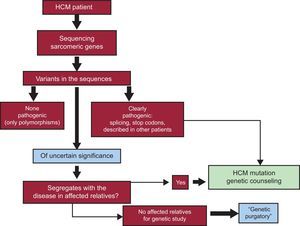
 can be considered the cause of the disease in the patient and carrier relatives. In the case of variants of uncertain significance (mainly amino acid changes) the mutation must prove to segregate with the disease, which means that several affected relatives should be available for testing. In the absence of affected family, the pathogenicity of many variants will remain undefined (“genetic purgatory”). HCM, hypertrophic cardiomyopathy.")
Genetic testing flowchart showing the steps from genetic assessment of a patient to genetic counseling in the family members. Variants that are clearly pathogenic (eg, by their effect, changes due to intronic splicing, introduction of premature stop codons) can be considered the cause of the disease in the patient and carrier relatives. In the case of variants of uncertain significance (mainly amino acid changes) the mutation must prove to segregate with the disease, which means that several affected relatives should be available for testing. In the absence of affected family, the pathogenicity of many variants will remain undefined (“genetic purgatory”). HCM, hypertrophic cardiomyopathy.
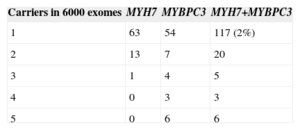
Until recently, we have been immersed in a genetic “age of innocence”, in which any amino acid change present in an HCM patient and not found in a control group (usually 100-500 individuals) was classified as a disease-causing mutation in that particular case. Monitoring was recommended in carrier members, and the future risk of developing the disease was considered low in noncarriers. If there were asymptomatic carriers of advanced age in the family, it would be concluded that the mutation had low penetrance, which would imply a benign character. In some families, HCM might develop in a member not carrying the mutation, which would indicate the presence of a second mutation in that individual. Over the last few years, however, information on genetic variation has been compiled in thousands of controls, and more than 5000 exomes have been sequenced (exomes, the gene sequences that code for proteins, account for around 5% of the three thousand million nucleotides). Nonetheless, when we search in these databases for the sarcomeric mutations implicated in HCM, we are faced with a harsh reality: many of them have been found in at least 1 of the controls (Table 2).37 At this point, doubts arise: Could this variant, formerly considered a disease-causing mutation, actually be a “rare” polymorphism unrelated to HCM?
Number of Carriers of “Rare” Nucleotide Variants That Would Give Rise to an Amino Acid Change, Present in Only 1 to 5 Exomes Sequenced
| Carriers in 6000 exomes | MYH7 | MYBPC3 | MYH7+MYBPC3 |
|---|---|---|---|
| 1 | 63 | 54 | 117 (2%) |
| 2 | 13 | 7 | 20 |
| 3 | 1 | 4 | 5 |
| 4 | 0 | 3 | 3 |
| 5 | 0 | 6 | 6 |
The term genetic purgatory was coined by J. Ackerman, a cardiologist in the Mayo Clinic, to refer to the situation of uncertainty that occurs when amino acid changes detected in a patient are also present in at least one of the control exomes sequenced.38 Although that author's article refers to gene changes in long QT syndrome, the concept can be extended to HCM and most other hereditary diseases. These changes are considered VUS; that is, they may or may not be the cause of a disease. This uncertainly leads to problems for genetic counseling. What is the point of analyzing these variants in the patient's family if their pathogenic status cannot be confirmed? Excluding the risk of developing the disease in noncarriers of a VUS would have consequences if there were another, unidentified mutation (in a different gene) and they were carriers of the mutation. Furthermore, recommending follow-up or even treatment in VUS carriers might be inappropriate as they might not have the culprit mutation (which, in reality, has not been identified). In the approximately 6000 exomes sequenced, there are 63 unique amino acid changes in MYH7 and 54 in MYBPC3 (Table 2). That is, 119 control individuals are carriers of a change that only occurred in each of them. This scenario becomes even more complex when the remaining frequently mutated sarcomeric genes are included (Table 1), which add up to 60 unique variants. If all these variants were pathogenic, 3% of the population would have a mutation in at least 1 of the 9 main genes implicated in HCM, and the estimated prevalence of the disease would be multiplied by 15 (1/500=0.2%).39 The situation would become even more muddled with the addition of other genes to the analysis, until a point would be reached when at least 1 “suspicious” change would almost certainly be found in a gene that (to a greater or lesser degree) had been linked with HCM.
Several bioinformatic algorithms are available to predict the pathogenicity of a sequence variant, and classification of an amino acid change as possibly damaging could favor its identification as a mutation. These programs use several criteria, and the degree of conservation of an amino acid between species is a criterion that carries considerable weight, following the principle that what is conserved through evolution must be functionally important and that any change at that level is likely to have consequences.40 Nonetheless, these are only predictions, and a variant classified as “probably pathogenic” may actually be benign.40 Indeed, if all the rare variants in sarcomeric genes classified as pathogenic by the PolyPhen-2 program were taken to be mutations, the prevalence of HCM would be much higher than the currently estimated value.41 This same scenario is seen in other cardiomyopathies such as dilated cardiomyopathy and right ventricular arrhythmogenesis. It should be stressed that genetic purgatory is a temporary stopover where we wait for someone to define these variants and release them from uncertainty.38 Some are surely pathogenic, but others are not, and until their association with the disease is clarified, we will have to tell patients and their families that “further evaluations may be needed and in the meantime we will maintain cardiologic monitoring”.
HOW TO ESCAPE FROM “GENETIC PURGATORY”Even when an amino acid change detected in a patient appears in the exome database, its pathogenic nature cannot be ruled out. It is always possible that the apparently healthy control carrier may have subclinical left-ventricular hypertrophy. It may be a young person and, if the variant has low penetrance, its effects may not be evident as yet. Even when exome data from a large number of healthy individuals of advanced age are collected, the situation could remain perplexing. In an analysis of the MYH7 and MYBPC3 genes in 300 individuals older than 70 years recruited in several health centers in Asturias (Spain) and showing no evidence of heart disease, unique variants were found in 9 participants (3%). Of course, echocardiography could be used to investigate left-ventricular hypertrophy in healthy control carriers of rare amino acid changes, as was done in the Framingham and Jackson Heart42 populations. More sensitive methods could even be used to seek evidence of the disease, such as magnetic resonance imaging.43
The percentages of VUS detected in HCM patients in the general population indicate that many of these rare variants must be pathogenic. In our series (n=444), 50 (11%) patients were carriers of amino acid changes unique to each, and within this total, 35 changes were not present in the exome database. If 11% of patients have rare variants vs 3% of the general population, many of these variants would necessarily have to be pathogenic. But, as was noted, we cannot conclude that a variant has a pathogenic effect solely because it is absent from the exome database; it could always be a rare polymorphism present in the particular patient.
How can we liberate VUS from this genetic purgatory? To be able to conclude that they are related to HCM, their segregation with the disease would have to be proven in the family of the index case, or alternatively, functional studies would have to be performed to demonstrate their pathogenic nature.
SegregationA pathogenic variant found in a patient should be present in all affected members of the patient's family. As mentioned earlier, mutations with a clear pathogenic effect will cause severe, early-onset forms of HCM; hence, it is very likely that there will be a family history of the disease and that various individuals will be available to confirm segregation. Furthermore, many of these variants have been found in several families and it is unlikely that they will be present in healthy controls and appear in exome databases. In contrast, it is more likely to detect VUS in patients with no family history of HCM and often diagnosed at an advanced age. Although these variants may be pathogenic, their penetrance would be low and they might be detected in controls, especially individuals younger than the age at which the symptoms manifest. Demonstration of pathogenicity by segregation with the disease follows a statistical rule; therefore, it depends on the number of affected individuals evaluated in a family and their degree of kinship.
BiochemistryA pathogenic variant should be “visible” by molecular assessment of the protein. For example, when a mutated gene is inserted in cultured cells, we should be able to see abnormal processing of the protein, with formation of aggregates and modifications in the cell phenotype. If a specimen of the patient's cardiac tissue is available, the protein should show the same abnormal pattern.
AnimalsThe pathogenic effect of a mutation can also be demonstrated by “creating” a transgenic animal (usually a mouse) that expresses the mutated protein and consequently should develop an HCM phenotype.44 This is a costly option and is currently unfeasible for all VUS.
“GET CRACKING”: SEEKING NEW GENES IN HYPERTROPHIC CARDIOMYOPATHYAlthough mutations in more than 20 genes are currently known to be associated with HCM, a high percentage of patients show no variants that could explain the presence of the disease.9,11,12 Again, we stress that this situation is more likely in patients with apparently sporadic disease and no clear family history of the condition. This implies that there must be other, still unidentified genes related to HCM, and the question is how to discover them among approximately 25 000 genes in the human genome. This task has been difficult to manage, even when it is limited only to genes encoding proteins that participate in cardiac physiology. However, it can now be accomplished by analyzing the exome, although interpretation of the results entails some difficulty.
First, it is assumed that hundreds of “possibly pathogenic” variants will be identified in various genes in the exome of a patient. Even when the search is limited to genes with cardiac relevance, various candidates will be identified. Once again, the pathogenicity of these variants will mainly be defined by their absence in controls, with the limitations this implies. When segregation of a possible mutation with HCM is lacking in a family, it may be impossible to demonstrate its relationship with the disease.
Based on these premises, to search for new genes, we would have to start with a patient showing no mutations in known HCM-related genes and have access to several affected family members who also harbor the possible mutation detected in the index case. Only a few families meet these criteria, as most HCM cases that might be caused by new genes are sporadic and no affected relatives will be available for genetic study. Ultimately, if there are many affected family members to verify the presence of the mutation, the number of candidate mutations/genes could be reduced to a few or even 1. In the specific case of HCM, few studies have focused on seeking new related genes by exome sequencing. Although many patients show no mutations known to be associated with the disease, there are very few families with several affected members to enable confirmation of a previously unknown mutation.
Once the candidate gene or genes are selected, functional assessment is performed to demonstrate that the mutation has an effect on the protein, or pathological evaluation is done on the patient's cardiac tissue to determine whether it shows changes. By adhering to this experimental approach, our group identified filamin C as a new mutated gene (FLNC) in HCM patients.45
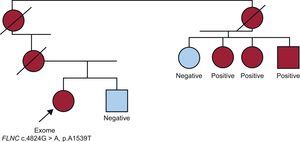
THE FILAMIN C CASEWe started with an HCM patient and various affected family members; DNA was available from 3 relatives (Figure 4). The patient tested negative for mutations in the main sarcomeric genes.12 Following sequencing of the patient's exome, more than 100 possible mutations were found in a number of genes; that is, alterations in DNA sequences that would give rise to amino acid changes that had not been described in controls in the exome database. We were able to narrow down the number of candidates to 25 by including only genes that encoded proteins related to cardiac physiology. After determining whether these variants were also present in the affected relatives, we found that only 1 change, c.C4824G in FLNC, which would result in an alanine to threonine change in the protein (p.A1539T), was present in all 4 affected individuals.45

Family in which FLNC was identified as the cause of hypertrophic cardiomyopathy. The index case was a woman with no mutations in the main sarcomeric genes. Following sequencing of her exome, all mutations known to be linked to hypertrophic cardiomyopathy were excluded. Among possible mutations in genes related to cardiac physiology, only the filamin C mutation was found in all the affected relatives.
Filamin C is a protein mainly expressed in striated muscle where it interacts with actin filaments and may function by communicating the cell membrane with the sarcomere.46 To verify that the amino acid change found had consequences on the function of the protein, a copy of the gene containing the change was created by directed mutagenesis and incorporated into rat cardiac myocytes, which were then cultured and submitted to functional analysis. In parallel, the same analyses were performed with cells transfected with a copy of FLNC having a normal sequence. These studies showed that FNLC with the possible mutation formed aggregates, whereas the normal form remained in the soluble cellular fraction.45 In addition, aggregates of other cytoskeletal proteins were observed, such as actin. Although the data showing family segregation with HCM and the cell studies clearly pointed to FLNC as the cause of the disease, a single finding in only 1 family could have limited value; ideally, the presence of these possible mutations should be demonstrated in other HCM patients. To this end, we sequenced FLNC in 92 patients with no mutations in the main genes implicated in HCM, and a possible mutation was found in 8 patients. Among these, 6 patients had at least 1 affected family member and all were carriers of the possible mutation.
Furthermore, 2 of the patients had undergone transplantation, and cardiac tissue was available for assessment. In both patients, histological evaluation showed intracellular aggregates of filamin C and sarcomeres with abnormal structure, displaying myofibrillar disorganization and fibrosis.45
The main conclusion of the study was that possible mutations in FLNC could be found in around 10% of HCM patients who did not have mutations in the main genes known to be associated with this disease. One might ask how confident we can be that these changes are actually mutations. The degree of confidence would be the same as for other HCM-related genes, as the basis is always an absence of the variants in controls and segregation with the disease in the family. Of course, some possible mutations in FLNC will be nonpathogenic changes, but if the frequency in our patients (9/93; 9.7%) is compared with that of unique variants that are possibly pathogenic in the exome database (91/6500; 1.4%), the statistics also favor the notion that FLNC is implicated in HCM. Irrespective of these considerations, the percentage of patients with possible mutations in FLNC will remain undefined until results from larger patient samples and from other populations are available.
A major problem encountered when assessing FLNC as a candidate in HCM is that the gene has already been related to familial forms of fibrillary myopathy (a condition affecting skeletal muscle), and some patients show cardiomyopathy in addition to the characteristic neuropathy.46–48 None of our patients with possible FLNC mutations had myopathy, and histological evaluation of muscle biopsy specimens in several of them excluded abnormalities.45 This phenomenon, in which mutations in a gene may give rise to heterogeneous clinical manifestations, is not unusual. For example, MYH7 has been related to a rare form of early-onset distal myopathy;49FHL1 mutations, which usually cause neuromuscular disease, have also been related to HCM with left ventricular diastolic dysfunction and no neuromuscular involvement50; and BAG3 mutations have been associated with early-onset neuropathy in various families, but also with dilated cardiomyopathy and no muscle involvement in others.51,52 As occurs in these genes/diseases, the available evidence indicates that mutations in different domains of filamin C may lead to clinical manifestations of myopathy or of HCM. Basic research is required to elucidate the molecular mechanisms by which FLNC is linked to HCM.
CONCLUSIONSMassive DNA sequencing has facilitated identification of the genetic bases of hereditary diseases by making genetic assessment accessible to a growing number of patients. In the case of HCM, when a bona fide mutation is found in a patient, this technique can be used for genetic counseling in the patient's relatives, identifying carriers that require monitoring, diagnosing new asymptomatic patients, and even in preimplantation genetic diagnosis. Massive sequencing has also disclosed the presence of many rare variants in the general population (healthy controls) to such a degree of genetic complexity that it could make interpretation of the results difficult in many patients. This is the case of patients with VUS, most of which involve amino acid changes that have not been found in other patients. In the absence of affected family members in whom segregation with the disease could be sought, these variants are classified as having uncertain significance because they may or may not be pathogenic (rare polymorphisms). Unless this dilemma is resolved, these variants cannot be used as a reliable basis for genetic counseling in the patient's family.
Massive sequencing also facilitates the search for mutations in new genes through analysis of the genome of HCM patients not carrying mutations in any of the genes known to be associated with the disease. Detection of VUS requires assessment of the new genes in HCM-affected family members, in whom the presence of these possible mutations should be confirmed.
CONFLICTS OF INTERESTNone declared.