Keywords
INTRODUCTION AND OBJECTIVES
Ventricular ejection generates highly pulsed flows and pressures in the arterial system. Blood vessels have structural and functional characteristics that reduce or buffer pulsatility to ensure continuous capillary flow which, in turn, optimizes tissue perfusion and prevents damage to fine capillary walls due to pressure waves from heart beats.1 Buffering by the arteries is also beneficial both for the heart because of the decrease in cardiac workload and myocardial tension, and for the great arteries because of the decrease of stress-induced fatigue of the artery walls.2
Several cardiovascular factors contribute to global buffering function (GBF) of the pressure waves and flow in pulmonary and systemic circuits. The great arteries are responsible for a component of this global capacity through their arterial wall buffering function (AWBF).1 This component can be analyzed with a Kelvin-Voigt spring-dashpot model,3,4 in which the spring corresponds to the arterial elasticity and the dashpot to arterial viscosity (viscoelastic model). Viscosity provides an indication of capacity to dissipate the energy of wave pulse components, whereas elasticity is a measure of energy storage.1,3,4
Arterial viscoelastic properties may vary because of chronic changes in the arterial wall structure or histological composition (arterial wall remodeling).5-9 Alternatively, acute responses to variations in pressure10 or arterial volume11 (passive changes) may occur. Acute responses may also be provoked by changes in the degree of activation of vascular smooth muscle (VSM) (active changes).3,4,8,12 Activation of aortic VSM allows intravascular pressure to increase while retaining arterial wall stiffness.3,4,10,12
Most work on buffering function and the effect of muscular activation has been done in systemic arteries. Results from this research cannot be applied to the pulmonary circuit because of hemodynamic and structural differences between the two circuits.12,13 We are aware of only a few studies that characterize the viscoelastic properties of the pulmonary artery, and fewer still that examine the pulmonary and systemic circuit at the same time in healthy animals.
The aim of this work was to characterize and compare the following in vivo: a) the viscoelastic properties of the aorta and the pulmonary artery and how they affect AWBF, and b) the direct effect of VSM activation on viscoelastic properties and AWBF.
MATERIALS AND METHOD
Instrumentation and surgery
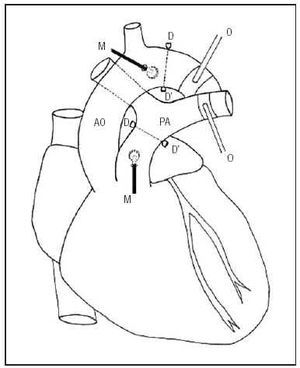
Six Merino sheep (26±4.5 kg) were anesthetized with intravenous sodium pentobarbital (35 mg/kg) and mechanical ventilation was established. The flow rate and respiratory rate were set to maintain pO2 above 85 mm Hg, pCO2 between 35 and 40 mm Hg, and pH between 7.35 and 7.4. The saphenous vein was catheterized to administer saline solution, the anesthetic and the vasoactive agent. A left-side thoracotomy exposed the great vessels. Pressure transducers (Konigsberg Instruments, Inc., Pasadena, CA) were placed in the ascending aorta and the primary pulmonary artery through minimal incisions in the vascular wall. A pair of ultrasound crystals (5 MHz, 3 mm diameter) were sutured distally to each pressure transducer. These were connected to a sonomicrometer (Triton Technology, San Diego, CA) to measure the distance between crystals (the speed of the ultrasound signal in these tissues is known to be 1580 m/s). Signal quality was optimized with an oscilloscope. The fast measurement time and linear response of the sensors ensured precise and reproducible measurements of the pressure and diameter.3,4,10,12 Pneumatic occluders were placed around the descending aorta and the left branch of the pulmonary artery. Figure 1 shows a schematic representation of the instrumentation.

Fig. 1. Schematic diagram of the experimental instrumentation. AO indicates aorta; PA, pulmonary artery; D-D', ultrasound crystals; M, pressure microtransducers; O, pulmonary and aortic pneumatic occluders.
Experimental protocol
The pressures and diameters were recorded under three experimental conditions:
1. Stable normotensive state (control).
2. Acute passive arterial hypertension (PH) induced by partial occlusion of the descending aorta and the left branch of the pulmonary artery for at least 7 seconds. Given the rapid increase in arterial pressure and diameter, arterial wall responses reflected passive changes in arterial wall viscoelastic properties.10,12 Occlusion lasted until mean arterial and systolic pressures similar to those in active hypertension were obtained.
3. Active arterial hypertension (AH) induced by continuous intravenous (i.v.) infusion of phenylephrine (Sigma, St. Louis, MO) at a dose of 5 µg/kg/min. Blood pressure was measured 15 minutes after starting drug administration to ensure pressure and diameter had stabilized.
After each maneuver, 10 minutes elapsed to allow pressure and diameter to return to control values. The animals were sacrificed at the end of the experiment. Necropsy confirmed the correct position of the pressure and diameter sensors. The surgical procedure was performed in accordance with international ethical standards and guidelines for research with experimental animals.14
Data collection and analysis
The pressure and diameter of the aorta and pulmonary artery were monitored in real time with hardware and software specially designed in our laboratory (SAMAY M16) at a sampling frequency of 200 Hz.12 The animal was disconnected from the ventilator at the end expiration for data acquisition because the viscoelastic properties of the pulmonary artery can vary during the ventilatory cycle.11 Both arteries were simultaneously monitored for 10 to 15 consecutive beats in each of the three experimental conditions. The area of hysteresis loop delineated by the arterial pressure-diameter curves (Figure 2) was minimized by increasing the values for viscosity index, which is proportional to the first derivative of arterial diameter (Annex). From the purely elastic pressure-diameter relationship thus obtained, elasticity was calculated by taking the first derivative of the mean diastolic pressure (Annex). The viscosity/elasticity coefficient was used to quantify AWBF, and GBF was estimated from a 2-element Windkessel model by fitting an exponential function to the diastolic tail of the arterial pressure curve (Annex).

Fig. 2. A: pulmonary pressure/diameter ratio in an animal under control conditions. B: aortic pressure/diameter ratio in an animal under control conditions.

Statistical analysis
Data were expressed as mean±SD (standard deviation). Statistical analysis was performed by ANOVA, followed by Student's t test for paired samples. Statistical significance was set at P<.05 for t and F values.
RESULTS
Table 1 shows the values for aortic and pulmonary hemodynamic variables under the three experimental conditions. For each condition, the pressures were higher (P<.05) and the diameters smaller (P<.05) in the aorta than in the pulmonary artery. The mean diastolic and systolic pressures in the two hypertensive conditions (PH and AH) were similar to each other but higher than those in control conditions for both arteries (P<.05). With the PH maneuver, mean systolic and diastolic pressures were greater than control values (P<.05) and those with AH (P<.05). With AH, pulmonary artery diameters were less than in the control (P<.05) but were similar to those in the aorta. Heart rate was similar in all three experimental conditions.

Table 2 shows the viscoelastic and AWBF values for the aorta and pulmonary artery, as well as the GBF for the systemic and pulmonary circuit in the three experimental conditions. Under each condition, elasticity and viscosity were always higher in the aorta then in the pulmonary artery (P<.05). No differences in viscosity were seen between control and PH conditions for either artery, but viscosity increased significantly during AH. Elasticity was lower during AH (P<.05) than during PH in both arteries. Elasticity during AH was significantly greater than control (P<.05) in the aorta only.

Arterial wall buffering function tended to increase in AH compared to the control condition, although the change was not significant either for the same artery or for the difference between arteries. In both arteries, AWBF during PH was lower than during the control condition and during AH (P<.05). Arterial wall buffering function values for the aorta and the pulmonary artery were similar under all different experimental conditions.
Systemic GBF was larger than pulmonary GBF (P<.05) under all experimental conditions. In each circuit, the GBF values were lower for PH (P<.05) than for control and AH, whereas for AH they were similar to control values. For all conditions, r2 was greater than 0.99, indicating that the exponential fit during calculation of GBF was good.
DISCUSSION
The aim of this work was to characterize and compare the following in vivo: a) the viscoelastic properties of the aorta and the pulmonary artery and how they affect AWBF, and b) the direct effect of VSM activation on viscoelastic properties and AWBF. We present a new index which is calculated from the arterial wall viscosity/elasticity coefficient4 to characterize the local buffering capacity of the arterial wall. The diastolic time constant is calculated to characterize the GBF for each circuit.
Basal viscoelastic properties
The mean pressure and arterial elasticity and viscosity were between 4.5 and 5.5 times higher in the aorta than the pulmonary artery under control conditions. These differences in the viscoelastic indexes may be due mainly to different pressures in each circuit.5,7,8 Given that both arteries have similar arterial wall components,6,15 factors that may explain the differences in the viscoelastic indexes are: a) different mechanical properties of the same arterial wall constituent; b) different net and relative proportions; c) different orientations of the arterial wall constituents; d) differences in the molecular conformation of the same material; e) differences in coupled functioning of the different arterial wall components, or f) differences in the net or relative quantity of extracellular matrix.8,16 With regard to elasticity, Keeley and Alatawi5 observed that arterial hypertension causes an increase in absolute quantities of aortic collagen and elastin, but the ratio between the two remains constant.5 This observation has prompted some investigators to propose a «quantum» or «unit» of elasticity that is determined by the ratio of collagen to elastin.17 The number of these «units» and therefore the degree of arterial wall elasticity would depend on the mean basal arterial pressure.
Arterial wall viscosity, on the other hand, is widely thought to depend on smooth muscle cells (SMC).7,8 An increase in blood pressure has been shown to correspond to proliferation of SMC.7,8,18,19 Collagen and elastin synthesis increases when more muscle cells are subjected to cyclic stretching or displacement.7 The higher pressure in the aorta compared to the pulmonary artery would lead to more SMC and therefore a higher basal viscosity and greater synthesis and deposit of elastic fibers, making the wall of the aorta more elastic. Another approach is to assume a fixed ratio of elastin, collagen and VSM in the arterial wall, which would explain why the viscosity/elasticity coefficient (AWBF) is similar for both arteries. This would reflect the basic functional contractile-elastic unit proposed by Davies,17 with a similar viscoelastic set point for both arteries.
Elasticity during arterial hypertension
Elasticity depends strongly on pressure and therefore increased significantly in both arteries during PH.3,4,10,12 Elastin, collagen, and VSM have different elastic moduli.3 They all start to stretch at different levels of arterial wall strain, thus the ratio of pressure to diameter is non-linear.3,10,12 At higher pressure, the recruitment of more rigid collagen fibers is greater, and elasticity increases.3,12
During AH, elasticity showed a similar tendency in both arteries, although elasticity increased significantly with respect to control values only in the aorta. The degree of activation of VSM also affected arterial elasticity. Elastin and collagen keep the level of arterial stress stable, which allows cyclic elastic arterial stretching and recovery but prevents over-distension and arterial wall disruption.3 In contrast, VSM is a dynamic component whose elasticity depends not only on the distension pressure but also on its extent of activation. Previous studies performed in systemic arteries3,4,10 and the pulmonary artery12 showed that activation of VSM increases arterial wall elasticity as a function of deformation or diameter (isometric analysis), or decreases as a function of strain or pressure (isobaric analysis). Our data agree with these studies because elasticity, when measured at constant pressure, decreased with muscular activation (PH vs AH). Isobaric contraction of the arterial diameter during AH might reduce the participation and hence also recruitment of collagen fibers, thus increasing arterial wall elasticity (as in PH).3,12 Unlike the aorta, elasticity in the pulmonary artery did not differ significantly between control and AH conditions, which may be due to differences in diameter during muscular activation. The diameter of the aorta during AH did not change with respect to the control value, but activation of VSM of the pulmonary artery decreased the arterial diameter with respect to the control condition, therefore less collagen was recruited.
Viscosity during arterial hypertension
Only the activation of VSM increased viscosity with respect to the control value in both arteries. Moreover, viscosity was greater in the aorta than in the pulmonary artery for all experimental conditions. Arterial wall viscosity has been associated mainly with energy loss between beats through the amount of VSM present and its degree of activation.3,7,8,10 The viscosity of arterial tissue might be explained by one of two theories. The «passive» theory assumes that viscosity is a property of the constituents of the arterial wall, the most active being VSM.3,7 The «active» theory considers the mechanisms generating muscle contraction (activation) and myogenic response to stretching to be important.7 Our work shows that under basal conditions, the arterial wall can be considered viscous, and that viscosity increases during muscular activation. Therefore, activation of pulmonary VSM modifies arterial wall elasticity and viscosity, as it does in systemic arteries.3,10,12
Arterial wall and global buffering functions
Arterial wall buffering function was defined by arterial wall viscosity/elasticity ratio in the present study. Elasticity and viscosity are greater in the aorta than in the pulmonary artery under control conditions, but the viscosity/elasticity coefficient was similar in both arteries. That is, absolute values of elasticity and viscosity may differ for each artery, but the buffering capacity of bot h arterial walls remains the same. The similar AWBF for the aorta and pulmonary artery indicate a functional adaptation of the great arteries to their particular pressure and flow conditions. The actual value of AWBF may therefore be an optimum value for local buffering. The viscosity/elasticity ratio would protect the wall from the high frequencies present in the pulse wave (stress fatigue)4 and would change towards the peripheral circulation as the histological structure of the artery changes.
Arterial wall buffering function decreased significantly in both arteries during PH because the arterial wall becomes more rigid. But during AH, AWBF did not change significantly with respect to the control value, indicating that VSM activation prevents a decrease in AWBF even though the arterial walls are subjected to higher pressures. Our data clearly show that arterial walls subjected to equal pressure may show different buffering and protection capacities. This also suggests that the size of the pressure pulse is not a suitable indicator of the functional state of the arterial wall.
To analyze the contribution of arterial wall buffering on global hemodynamics, the GBF for each circuit was calculated during the same beats. Global buffering function characterizes the ability of each vascular circuit to buffer the pressure wave, and is determined mainly by overall compliance and overall peripheral resistance of the circuit.4
Global buffering function was always higher in the systemic circuit than in the pulmonary one despite similar values for AWBF in both arteries. Higher systemic GBF may be due partly to higher peripheral resistance and the longer circuit.13 In both arteries, GBF decreased significantly during PH with respect to the control value. An isobaric comparison of GBF for AH and PH showed that muscular activation led to higher GBF. Global buffering function therefore behaved like AWBF, which might indicate that activation of the arterial wall muscle changes the buffering capacity, thus helping to generate a greater overall buffering effect in both circuits. Unlike AWBF, GBF is significantly greater in the systemic circuit, probably because more effective filtering is required by the larger number of harmonics with larger amplitudes generated by the left ventricle.20
If GBF and AWBF are kept constant, VSM activation can be beneficial during hypertension in three ways. First, it favors ventricular-arterial coupling by keeping the arterial impedance constant. (Impedance is inversely proportional to vascular cross-section and directly proportional to arterial wall stiffness.) Second, it maintains local protection in the great arteries by reducing stress fatigue in the arterial walls. Finally, activation ensures continuous low-pressure flow in the microcirculation.
The overall control mechanisms of the arterial system act mainly through variations in overall peripheral resistance and total compliance, and modify the diastolic decay time constant. A local control mechanism may therefore act by modifying arterial wall viscosity, which is dependent on VSM activation, which in turn would modulate the local arterial wall time constant. Support for the role played by the endothelium in local regulation comes from several studies that show that the endothelium has an active function in vascular remodeling and in the control of viscoelastic properties of the vascular wall.18 Future research will need to characterize the «overall control-to-local control» relationship in the arterial system in integrated cardiovascular responses, both in physiological and pathophysiological situations.
Clinical implications
From a clinical point of view the pulse pressure refers only to the pulse component of arterial pressure, but it is actually a complex parameter that depends on the ventricular ejection pattern, GBF, the retrograde wave and arterial stiffness.21 This would explain the similar increase in pulse pressure in both arteries in the two types of hypertension. Therefore, analysis of pulse pressure alone is not enough to distinguish changes in arterial wall stiffness that are secondary to arterial pressure or arterial wall mechanics (AWBF), and does not provide information on the consequences for ventricular-arterial coupling. Modern high-resolution echocardiographic techniques (echo-tracking) will allow continuous non-invasive measurement of systolic variations in the diameter of the aorta and pulmonary arteries in humans.22 If arterial pressure is recorded simultaneously, characterization of parietal elasticity and viscosity will be possible from the pressure-diameter loop.
CONCLUSIONS
Viscoelasticity and overall buffering are greater in the main artery of the systemic circulation than in the pulmonary circulation. However, AWBF is similar in the aorta and pulmonary artery. The elasticity of both arteries is highly dependent on blood pressure, and viscosity values related to the amount of vascular smooth muscle and its degree of activation. During passive hypertension, AWBF and GBF decrease significantly. Activation of the vascular smooth muscle has beneficial effects on arterial circulation because AWBF and GBF values recover through a decrease in elasticity and an increase in viscosity, despite the increase in arterial pressure.
ACKNOWLEDGEMENTS
The authors thank Mr Elbio Agote and Ms Edith Moraes for technical assistance, as well as Ismael Aguirre BSc for the figure.
Correspondence:
Dr. D. Bia.
Departamento de Fisiología. Facultad de Medicina.
General Flores, 2125. 11800 Montevideo. Uruguay.
E-mail: dbia@fmed.edu.uy