


Pulmonary arterial hypertension (PAH) is characterized by increased pulmonary vascular resistance, right ventricular dysfunction and death. Despite scientific advances, is still associated with high morbidity and mortality. The aim is to describe the clinical approach and determine the prognostic factors of patients with PAH treated in a national reference center over 30 years.
MethodsThree hundred and seventy nine consecutive patients with PAH (January 1984 to December 2014) were studied. Were divided into 3 periods of time: before 2004, 2004-2009 and 2010-2014. Prognostic factors (multivariate analysis) were analyzed for clinical deterioration.
ResultsMedian age was 44 years (68.6% women), functional class III-IV: 72%. An increase was observed in more complex etiologies in the last period of time: Pulmonary venooclusive disease and portopulmonary hypertension. Upfront combination therapy significantly increased (5% before 2004 vs 27% after 2010; P < .05). Multivariate analysis showed prognostic significance in age, sex, etiology and combined clinical variables as they are independent predictors of clinical deterioration (P < .05). Survival free from death or transplantation for the 1st, 3rd and 5th year was 92.2%, 80.6% and 68.5% respectively. The median survival was 9 years (95% confidence interval, 7.532-11.959)
ConclusionsThe PAH is a heterogeneous and complex disease, the median survival free from death or transplantation in our series is 9 years after diagnosis. The structure of a multidisciplinary unit PAH must adapt quickly to changes that occur over time incorporating new diagnostic and therapeutic techniques.
Keywords
Pulmonary arterial hypertension (PAH) is defined as a group of diseases characterized by a progressive increase in pulmonary vascular resistance that leads to right ventricular failure and death. Prognosis is determined by the pathophysiological interaction between the rate of progression of obstructive changes in the pulmonary microcirculation and the adaptive response of the right ventricle. The main known prognostic factor in this disease is the extent of right ventricular dysfunction.
Pulmonary arterial hypertension is a rare disease with an estimated prevalence in different registries of between 15 and 26 cases per million population older than 14 years.1 Thanks to the scientific community's efforts, more than 30 multicenter clinical trials have been designed and conducted,2 which have allowed the development of 5 drug classes: prostacyclin analogues, phosphodiesterase-5 inhibitors, guanylate cyclase stimulators, prostacyclin receptor agonists, and endothelin receptor antagonists. These drugs, along with diagnostic and prognostic advances, have revolutionized PAH treatment, as reflected in 3 clinical practice guidelines that encompass the accumulated scientific evidence, published in 2004, 2009, and 2015, respectively.3–5
Previously, lung transplantation (LT) was the only treatment available, but following the discovery of various specific drugs, LT is now the final treatment option for patients who do not respond to pharmacological therapy.3 Despite advances, PAH remains a disease with a high associated morbidity and mortality, with a 5-year survival of 65% in Spain.1
The aim of our study was to describe the changes in therapeutic strategies and determine the prognostic factors and long-term survival of a cohort of patients with a diagnosis of PAH, in a national referral center, over a period of 30 years.
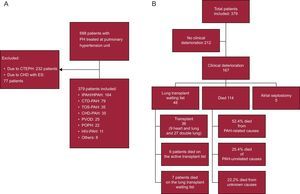
METHODSStudy Design and PopulationThis was an ambispective observational cohort study of patients diagnosed with group 1 PAH (idiopathic, familial, or hereditary PAH, forms associated with connective tissue disease, human immunodeficiency virus [HIV], portal hypertension, rapeseed oil, congenital heart disease, pulmonary veno-occlusive disease, and other less common causes [Osler-Weber-Rendu and hemolytic anemia]) from January 1984 to December 2014, who were treated at the Pulmonary Hypertension Multidisciplinary Unit of the Hospital Univeristario 12 de Octubre. The Hospital Universitario 12 de Octubre Ethics Committee approved the study. Patients seen from the year 2000 onwards were prospectively included in the database; those seen prior to the year 2000 were retrospectively included. Date of diagnosis was defined as the first right heart catheterization performed. We excluded congenital heart diseases with Eisenmenger syndrome and chronic thromboembolic pulmonary hypertension (Figure 1).

Flow diagram of patients and clinical deterioration. A: distribution of patients. B: distribution of clinical deterioration. CHD, congenital heart disease; CTD, connective tissue disease; CTEPH, chronic thrombo-embolic pulmonary hypertension; ES, Eisenmenger syndrome; HIV, human immunodeficiency virus; HPAH, hereditary pulmonary artery hypertension; IPAH, idiopathic pulmonary artery hypertension; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; POPH, portopulmonary hypertension; PVOD, pulmonary veno-occlusive disease; TOS, toxic oil syndrome.
Patients were divided according to 3 time intervals, to coincide with the guidelines available for each period (European Society of Cardiology/European Respiratory Society guidelines) as follows:
- •
Diagnosis prior to 1 January 2004.
- •
From 1 January 2004 to 31 December 2009 (2004 guidelines).4
- •
From 1 January 2010 to 31 December 2014 (2009 guidelines).5
Diagnosis of PAH was based on a diagnostic algorithm and the hemodynamic criteria recommended in the guidelines from each period.
The following variables were analyzed at diagnosis: demographic data, PAH etiology, functional class (FC), 6-minute walk test (6MWT), right atrial pressure, cardiac output, mean pulmonary artery pressure, pulmonary vascular resistance, pericardial effusion, and initial treatment. The event “clinical deterioration” was defined by the first event to occur: death, inclusion on the LT waiting list, or atrial septostomy.
Statistical AnalysisDescriptive results are presented as frequency and percentage for qualitative variables and mean ± standard deviation or median [interquartile range] for quantitative variables. To calculate the statistical significance (P < .05) of comparisons for qualitative variables between groups, the Fisher exact test was used. For quantitative variables with normal distribution, we used the Student t test, and analysis of variance for independent groups when 2 or more groups were being compared. Nonparametric tests were used when the variable did not follow a normal distribution.
To identify the parameters that were “predictors of clinical deterioration”, bivariate analysis was performed for each variable regarding the time until deterioration; Kaplan-Meier curves and the log-rank test were used to compare the curves. Subsequently, those variables that were statistically significant (P < .05) in the bivariate analyses were selected and included in the multivariate Cox regression model for time until deterioration.
To determine if etiology was a prognostic factor, we grouped etiologies according to the median survival obtained on the Kaplan-Meier curves such that there were no significant differences between the etiologies forming each group. The first group comprised idiopathic PAH/hereditary PAH/rapeseed oil-associated PAH/congenital heart disease-associated PAH and HIV-associated PAH (median survival, 11.9 years, 95% confidence interval [95%CI], 8.4-18.1 years); the second group comprised PAH associated with connective tissue disease/portopulmonary hypertension/other (median survival, 5.9 years; 95%CI, 4.4-8.1 years); and group 3 comprised those with PVOD (median survival, 2.5 years; 95%CI, 1.2-7.1 years). The Kaplan-Meier graphs of this analysis are available in the .
Taking into account the most recent clinical practice guidelines,6 we established composite criteria as predictors of clinical deterioration, which combined FC and 6MWT distance or right atrial pressure. The criteria were defined as follows: criterion 1, FC III or IV with 6MWT < 475 m; criterion 2, FC III or IV with right atrial pressure > 8 mmHg. Finally, variables with a P-value < .05 in the multivariate model were selected as prognostic factors. The final result shows the relative risk (hazard ratio [HR]) and 95%CI. The C-index was used to calculate the concordance rate between the multivariate model prediction and the observed result for the whole sample.
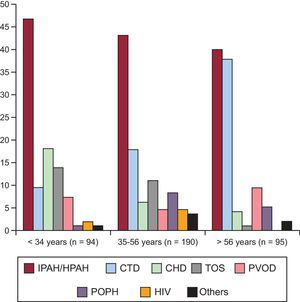
ResultsOf a total of 688 patients seen in our unit, 379 met the inclusion criteria for analysis (Figure 1A). Figure 1B shows the breakdown of clinical deterioration. Table 1 shows the main characteristics at diagnosis. Table 2 shows the differences by time period. The distribution of etiologies by diagnosis period shows a constant presence of idiopathic/hereditary PAH over time; a concentration of rapeseed oil-associated PAH is seen prior to 2004, as well as an increase in PVOD, congenital heart disease-associated PAH, and portopulmonary hypertension over time. The distribution of etiologies by age percentile (Figure 2) shows a decrease in idiopathic/hereditary PAH, congenital heart disease-associated PAH, and rapeseed oil-associated PAH as age increases. In patients older than 56 years, an increase was noted in forms associated with connective tissue disease (92% scleroderma). Genetic study was introduced in 2011, and genetic mutations were detected in 65% of familial forms of the disease and 18% of cases of sporadic idiopathic PAH. All patients with familial PVOD had a mutation in the eukaryotic initiation factor-2 kinase A4 (EIF2KA4) gene.
Baseline Characteristics
| Patients, no. | 379 |
| Sex | |
| Female | 260 (69) |
| Clinical classification of PAH | |
| Congenital heart disease | 33 (9) |
| Connective tissue disease | 79 (21) |
| PVOD | 25 (7) |
| Portopulmonary hypertension | 22 (6) |
| Idiopathic | 164 (43) |
| Toxic oil | 35 (9) |
| HIV | 11 (3) |
| Other etiologies | 10 (3) |
| Age at diagnosis, y | 44 [34-56] |
| Follow-up time, y | 5 [2-8] |
| 6MWT, meters | 396 [325-475] |
| Hemodynamic parameters | |
| Mean pulmonary artery pressure, mmHg | 56 ± 15 |
| Right atrial pressure, mmHg | 9 ± 5 |
| PVR, WU | 13 ± 7 |
| CI (L/min/m2) | 2 ± 0.8 |
| Functional class | |
| I-II | 105 (28) |
| III-IV | 274 (72) |
| Echocardiography | |
| Pericardial effusion | 54 (14) |
| Type of treatment | |
| Monotherapy | 321 (85) |
| Dual therapy | 42 (11) |
| Triple therapy | 6 (2) |
6MWT, 6-minute walk test; HIV, human immunodeficiency virus; PAH, pulmonary arterial hypertension; PVOD, pulmonary veno-occlusive disease; PVR, pulmonary vascular resistance; WU, Wood Units.
Unless otherwise indicated, data are expressed as n (%), mean ± standard deviation or median [interquartile range].
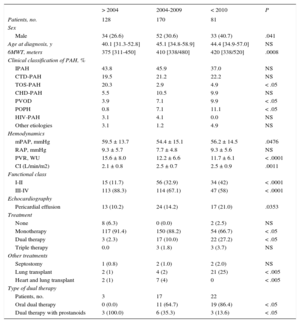
Baseline Characteristics by Time Period of Diagnosis
| > 2004 | 2004-2009 | < 2010 | P | |
|---|---|---|---|---|
| Patients, no. | 128 | 170 | 81 | |
| Sex | ||||
| Male | 34 (26.6) | 52 (30.6) | 33 (40.7) | .041 |
| Age at diagnosis, y | 40.1 [31.3-52.8] | 45.1 [34.8-58.9] | 44.4 [34.9-57.0] | NS |
| 6MWT, meters | 375 [311-450] | 410 [338/480] | 420 [338/520] | .0008 |
| Clinical classification of PAH, % | ||||
| IPAH | 43.8 | 45.9 | 37.0 | NS |
| CTD-PAH | 19.5 | 21.2 | 22.2 | NS |
| TOS-PAH | 20.3 | 2.9 | 4.9 | < .05 |
| CHD-PAH | 5.5 | 10.5 | 9.9 | NS |
| PVOD | 3.9 | 7.1 | 9.9 | < .05 |
| POPH | 0.8 | 7.1 | 11.1 | < .05 |
| HIV-PAH | 3.1 | 4.1 | 0.0 | NS |
| Other etiologies | 3.1 | 1.2 | 4.9 | NS |
| Hemodynamics | ||||
| mPAP, mmHg | 59.5 ± 13.7 | 54.4 ± 15.1 | 56.2 ± 14.5 | .0476 |
| RAP, mmHg | 9.3 ± 5.7 | 7.7 ± 4.8 | 9.3 ± 5.6 | NS |
| PVR, WU | 15.6 ± 8.0 | 12.2 ± 6.6 | 11.7 ± 6.1 | < .0001 |
| CI (L/min/m2) | 2.1 ± 0.8 | 2.5 ± 0.7 | 2.5 ± 0.9 | .0011 |
| Functional class | ||||
| I-II | 15 (11.7) | 56 (32.9) | 34 (42) | < .0001 |
| III-IV | 113 (88.3) | 114 (67.1) | 47 (58) | < .0001 |
| Echocardiography | ||||
| Pericardial effusion | 13 (10.2) | 24 (14.2) | 17 (21.0) | .0353 |
| Treatment | ||||
| None | 8 (6.3) | 0 (0.0) | 2 (2.5) | NS |
| Monotherapy | 117 (91.4) | 150 (88.2) | 54 (66.7) | < .05 |
| Dual therapy | 3 (2.3) | 17 (10.0) | 22 (27.2) | < .05 |
| Triple therapy | 0.0 | 3 (1.8) | 3 (3.7) | NS |
| Other treatments | ||||
| Septostomy | 1 (0.8) | 2 (1.0) | 2 (2.0) | NS |
| Lung transplant | 2 (1) | 4 (2) | 21 (25) | < .005 |
| Heart and lung transplant | 2 (1) | 7 (4) | 0 | < .005 |
| Type of dual therapy | ||||
| Patients, no. | 3 | 17 | 22 | |
| Oral dual therapy | 0 (0.0) | 11 (64.7) | 19 (86.4) | < .05 |
| Dual therapy with prostanoids | 3 (100.0) | 6 (35.3) | 3 (13.6) | < .05 |
6MWT, 6-minute walk test; CHD, congenital heart disease; CI, cardiac index; CT, connective tissue disease; HIV, human immunodeficiency virus; IPAH, idiopathic pulmonary artery hypertension; mPAP, mean pulmonary artery pressure; NS, not significant; PAH, pulmonary arterial hypertension; POPH, portopulmonary hypertension; PVOD, pulmonary veno-occlusive disease; PVR, pulmonary vascular resistance; RAP, right atrial pressure; TOS, toxic oil syndrome; WU, Wood units;.
Unless otherwise indicated, data are expressed as no. (%), mean ± standard deviation, or median [interquartile range].

Distribution of patients by age percentile. CHD, congenital heart disease; CTD, connective tissue disease; HIV, human immunodeficiency virus; HPAH, hereditary pulmonary artery hypertension; IPAH, idiopathic pulmonary arterial hypertension; PAH, pulmonary arterial hypertension; POPH, portopulmonary hypertension; PVOD, pulmonary veno-occlusive disease; TOS, toxic oil syndrome.
Initial treatment with prostanoid monotherapy has decreased over time; since the introduction of oral therapy, it has been restricted to patients with a high risk profile. Patients were treated with prostacyclins for a median of 7.9 years [3.8-13.2 years] before clinical deterioration (table 2). Initial combined treatment increased (2.3%, 10%, and 27.2% in the first, second, and third periods, respectively) (P < .05).
Forty-eight patients were put on the transplant waiting list, and 36 transplants were performed: 9 heart and lung and 27 double lung transplants (Figure 1B). Of the patients included on the transplant waiting list, 22%, 43%, and 35% were diagnosed in the first, second, and third period, respectively (P < .05). All patients were treated with systemic prostacyclins prior to their inclusion on the transplant waiting list (except patients with PVOD). The number and type of transplant varied for each period: 14% (2 heart and lung and 3 double lung), 17% (5 heart and lung and 1 double lung), and 69% (all double lung) of all transplants occurred in the first, second, and third period, respectively. Lung transplantation has been performed in our hospital since 2011, and 1 patient with biventricular dysfunction was referred to another hospital for assessment for heart and lung transplant and was still on the waiting list at the end of our study follow-up.
Survival Analysis and Prognostic FactorsMedian follow-up was 4.5 years [2.2-8.2 years] and 14 patients (4%) were lost to follow-up. Figure 3 contains the variables that showed a significant difference in clinical deterioration in the bivariate analysis and were later included in the multivariate analysis. Survival free from clinical deterioration was higher in women, patients younger than 56 years, patients with right atrial pressure < 8 mmHg, with 6MWT > 475 m, and those in FC I-II (Figure 3). In terms of PAH etiology, group 3 had the worst survival (Figure 3C). The median survival free from transplant or death was 9 years (95%CI, 7.532-11.959 years). Figure 4A shows the data on survival free from clinical deterioration (91.9%, 80.1%, and 67.6% at the first, third, and fifth year, respectively) with a median survival of 8.2 years (95%CI, 7.0-11.6 years). Figure 4B shows the survival free from death or transplant in the first, third, and fifth year, which were 92.2%, 80.6%, and 68.5%. Cause of death was PAH-related in 52.4% (79% of these heart failure and 21% sudden death), in 25.4% it was unrelated to PAH (18% infections and 12% neoplasia), and in 22.2% cause of death was undetermined. Table 3 shows the multivariate analysis of prognostic factors for clinical deterioration. The C-statistic for the multivariate model was 0.8 (95%CI, 0.66-0.91).

. B: survival free from death or transplant. OS, overall survival.")
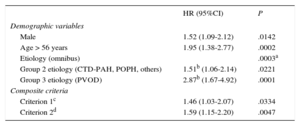
Multivariate Analysis: Prognostic Factors for Clinical Deterioration at the Time of Diagnosis
| HR (95%CI) | P | |
|---|---|---|
| Demographic variables | ||
| Male | 1.52 (1.09-2.12) | .0142 |
| Age > 56 years | 1.95 (1.38-2.77) | .0002 |
| Etiology (omnibus) | .0003a | |
| Group 2 etiology (CTD-PAH, POPH, others) | 1.51b (1.06-2.14) | .0221 |
| Group 3 etiology (PVOD) | 2.87b (1.67-4.92) | .0001 |
| Composite criteria | ||
| Criterion 1c | 1.46 (1.03-2.07) | .0334 |
| Criterion 2d | 1.59 (1.15-2.20) | .0047 |
6MWT, 6-minute walk test; 95%CI, 95% confidence interval; CTD, connective tissue disease; FC, functional class; HIV, human immunodeficiency virus; HR hazard ratio; PAH, pulmonary arterial hypertension; POPH, portopulmonary hypertension; PVOD, pulmonary veno-occlusive disease; RAP, right atrial pressure.
Our study is the first published series that reflects changes in PAH treatment over a 30-year period. It includes a consecutive cohort of patients with PAH treated in a national referral center with a high patient volume and provides information on survival > 5 years. Over the 3 decades, the causes of PAH have changed. Prior to 2004, there was a concentration of rapeseed oil-associated PAH, and of note is the diagnosis of new cases of this etiology in the most recent period, a long time after exposure. There was concentration of more complex patients after 2010; the most notable changes were the increases in PVOD and portopulmonary hypertension. These changes have created new demands, and with them has come the necessary adaptation of the pulmonary hypertension unit, which now offers portopulmonary hypertension screening in patients who are candidates for liver transplant, screening for scleroderma-associated PAH, genetic study in our population, and the transplant program, which started in 2011. Earlier diagnosis has been made possible due to the multidisciplinary work on screening patient populations with diseases associated with PAH and relatives of patients with hereditary forms of the disease. After 2004, the hemodynamic severity decreased and the FC at the time of diagnosis improved. Thus we found, as has been the experience of other groups,7,8 that early diagnosis and early intervention in PAH can translate to better long-term results.
Idiopathic Pulmonary Arterial HypertensionThis was the most common etiology. The median age at diagnosis of 44 years remained unchanged over time. In contrast, the COMPERA registry8 (2007-2012) and the United Kingdom registry9 (2001-2009) showed a median age at diagnosis of 71 and 62 years, respectively. This could be explained by fewer referrals of older patients to our hospital in recent years, related to the greater number of PAH units in Spain.
The median survival free from death or transplant was 11 years, longer than the 7 years reported by the REVEAL registry.6 Advanced age, FC III or IV, and the distance achieved in the 6MWT have been reported as established prognostic factors in patients with idiopathic PAH.6 In our study, the patients were younger, had better FC, and achieved a longer distance in the 6MWT, and prostacyclin use was greater; these factors could have contributed to the observed survival differences.
Connective Tissue Disease-associated Pulmonary Arterial HypertensionIn our study, 50% of patients older than 56 years had connective tissue disease-associated PAH; 92% of these had scleroderma. In the REVEAL registry,10 as in our series, patients with scleroderma had a mean age of 62 years and were the most prevalent group among patients older than 50 years. In patients with scleroderma-associated PAH in our series, survival in the first year was 81%, comparable to that described in the REVEAL registry.10 This one-year mortality close to 20%—which should act as a red flag to the medical community—led to the proposal of new screening11 and treatment protocols; our unit implemented these last year. Early diagnosis is key to improve survival.12
Rapeseed Oil-associated Pulmonary Arterial HypertensionThe toxic oil syndrome epidemic related to the ingestion of denatured rapeseed oil occurred in 1981 and affected around 20 000 people. Approximately 20% of those affected developed PAH, with apparent complete spontaneous remission in many of them; a small percentage developed a chronic severe form.1 Our hospital centralized the follow-up of patients with this type of PAH; 74% of the cases were diagnosed prior to 2004. However, new diagnoses continue to be made, some of which have intervals between exposure and PAH onset of more than 30 years. Such latency periods of variable duration are also described in the development of PAH after ingestion of anorexigens: in a series from the French registry, 44% of patients13 were diagnosed more than 5 years after exposure to the drug. Therefore, a history of ingesting rapeseed oil will be present in our setting and must be identified as a risk factor particular to Spain, despite the long latency period.
Congenital Heart Disease-associated Pulmonary HypertensionPatients with Eisenmenger syndrome were excluded because this has a different natural history and treatment. More than half of the rest of the patients had forms associated with systemic-pulmonary shunt repairs done in childhood, and the diagnosis of PAH had been made in the fourth decade of life. The REHAP registry14 describes that 25% of the 240 patients with congenital heart disease-associated PAH presented pulmonary hypertension following repair and, as in our series, survival was comparable to that of idiopathic PAH.
The second most common clinical group was that associated with restrictive defects (mostly interatrial communication), with a survival comparable to idiopathic PAH. In patients who received a LT for simple congenital heart disease-associated PAH with pulmonary-systemic shunt, the defect was closed during LT. Our unit is a national pioneer in the repair of congenital heart disease at the time of transplant, as recommended in the current transplantation guidelines.15
Pulmonary Veno-occlusive DiseasePulmonary venooclusive disease is characterized by rapid progression and a lack of treatment options. Mutations in the eukaryotic initiation factor-2 kinase A4 (EIF2KA4) gene have been associated with PVOD development, with an autosomal recessive inheritance pattern. Our group recently discovered a founder mutation (c.3344C>T [p.P1115L]) in said EIF2AK4 gene in 5 families of Romani ethnicity with PVOD characterized by disease onset at < 35 years old, consanguinity, low carbon monoxide diffusion capacity, and poor survival.16
In our series, 75% of patients with PVOD arrived at our hospital in a critical status, and 50% of them required support with an extracorporeal membrane oxygenator in the first 3 months after diagnosis, with no deaths while on the transplant waiting list. Other groups have described a high waitlist mortality rate in these patients, up to 22% at 6 months.17 The fast progression from time of diagnosis and lack of specific treatment mean that immediate referral is essential when PVOD is suspected, so that assessment by the transplant team may begin. In hereditary forms, a genetic study and screening of first degree relatives should be performed3 and heterozygous carriers should be identified. Medical practitioners in Spain should be particularly alert to this possibility and the diagnosis should not be delayed if there is clinical suspicion in patients from this ethnic group.
ProstacyclinsProstacyclins were used in 205 patients (56%). Seventy percent of those who died and 75% of patients in FC IV were receiving prostacyclin treatment. In the REVEAL registry, prostacyclin use was drastically lower and only 43% of those who died were receiving this treatment.18 The REHAP registry showed that from a national perspective, prostacyclins were underused, with only 50% of patients in FC IV receiving prostacyclins.2 This difference between data from national registries and an expert center can be explained by the need for a complex care organization that supports the use of these drugs and guarantees their treatment safety and excellence. Trained nursing staff is essential, as is a structured health education program and a 24-hour immediate response system (both in-person and telephone).
TransplantationInclusion on the LT waiting list depends on multiple factors, essentially the speed of progression of the disease and the scarcity of organs that leads to an ever-increasing waiting list time. Therefore, the latest clinical guidelines3 differentiate between 2 aspects: a) referral for LT assessment, which should be done early before all therapeutic possibilities have been exhausted, and b) active inclusion in the LT program. It is recommended to develop strategies as a bridge to transplant to minimize waiting list mortality. In our unit, after 2011, 15% of LTs for PAH were performed as an emergency and half needed a bridge to transplant strategy, essentially extracorporeal membrane support in 4 patients. In 5 patients, atrial septostomy allowed clinical stabilization of the patient and successful elective LT.19
Factors Predictive of RiskThe nonmodifiable demographic variables with a prognostic effect highlight the need to establish therapeutic strategies from the outset for patients with a high risk profile. In our series, the modifiable prognostic variables related to disease progression, such as FC III-IV, 6MWT < 475 m, and right atrial pressure > 8 mmHg (included in the composite criteria), were comparable to most other registries1,6 and reinforce the recommendation from the clinical guidelines to perform targeted assessments.5
An analysis was also performed with composite variables that distinguished patients in FC III-IV from those with higher risk who may need more aggressive treatment. Kane et al.20 explored this strategy in a cohort of 484 patients; the multifactorial prognostic estimation obtained a model with good predictive capacity, with a C-index of 0.84. Our multivariate model, with the inclusion of composite criteria (FC+6MWT and FC+right atrial pressure) had a C-index of 0.80, which highlights the importance of multifactorial evaluation in the prognostic estimation, as recommended in the current clinical practice guidelines.5
LimitationsThis was an ambispective observational study (12% of the patients were included retrospectively) of the experience of one center with a cohort of patients studied over a long period of time, with marked differences in each period in the etiology of PAH and the therapeutic options available. The analysis of risk factors and survival must be interpreted with caution, as there is a temporal bias. However, we think that a description of the clinical reality observed in a referral center over 3 decades gives a comprehensive view of the disease over time, and can help establish strategies aimed at improving the management of this disease. As this was a 30-year series, we did not use the newer tools for prognostic stratification available from 2010, namely N-terminal-pro-brain-natriuretic peptide, ergospirometric parameters, and the newer imaging techniques.
ConclusionsOur series presents the largest cohort of patients with PAH and the longest follow-up time in Spain described to date. The treatment of this disease has undergone drastic changes that allow early diagnosis in at-risk populations and highly complex treatment, including combined pharmacological therapy, extracorporeal membrane oxygenator, and LT. This study expands the information available on long-term survival patterns and provides new information on composite clinical variables and the usefulness of integrating these variables into the estimation of risk of clinical deterioration.
FUNDINGAn unconditional educational grant from ACTELION. The funding for this study was provided by the Instituto de Salud Carlos III and the Ministry of Economics and Competitiveness via the biomedical research center network (CIBER) for cardiovascular disease (CB16/11/00502).
CONFLICTS OF INTEREStP. Escribiano received an unconditional educational grant from Actelion for the statistical study performed by SAIL (statistical analysis biometrics); she has been a consultant for Actelion, Bayer, Pfizer, and GSK; she has received grants from Actelion and GSK, remuneration from Actelion, Bayer, Pfizer and GSK for lectures given, and remuneration from GSK for educational presentations given. P.E. Carreira has received a grant from ISCIII-FIS (Carlos III Health Institute, Healthcare Research Fund) and remuneration from Actelion for lectures given. J.F. Delgado Jiménez has received remuneration from Actelion and Bayer for lectures given. M.Á. Gómez Sánchez has been a consultant for Actelion, MSD, Ferrer, GSK, and Bayer, and has received remuneration from Actelion, MSD, Ferrer, GSK, and Bayer for lectures and educational presentations given. The rest of the authors declare no conflicts of interest.
- –
The epidemiological data provided by the large PAH registries have allowed investigators to establish prognostic factors. Current clinical practice guidelines recommend periodic multifactorial assessment of the disease. Survival, according to data from the Spanish PAH registry, is 65% at 5 years.
- –
Our study reflects the changes in the epidemiology and treatment of PAH over a 30-year period with a cohort of consecutive patients with PAH treated in a national referral center with a high patient volume. The multivariate analysis and the composite variables confirm the need for a follow-up based on periodic multifactorial assessment of the disease. The study provides information on survival at > 5 years of follow-up.
Xavier Masramón, of SAIL Biometría.