Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterized by progressive fibrofatty replacement of predominantly right ventricular myocardium. This cardiomyopathy is a frequent cause of sudden cardiac death in young people and athletes. The aim of our study was to determine the incidence of pathological or likely pathological desmosomal mutations in patients with high-risk definite ARVC.
MethodsThis was an observational, retrospective cohort study, which included 36 patients diagnosed with high-risk ARVC in our hospital between January 1998 and January 2015. Genetic analysis was performed using next-generation sequencing.
ResultsMost patients were male (28 patients, 78%) with a mean age at diagnosis of 45 ± 18 years. A pathogenic or probably pathogenic desmosomal mutation was detected in 26 of the 35 index cases (74%): 5 nonsense, 14 frameshift, 1 splice, and 6 missense. Novel mutations were found in 15 patients (71%). The presence or absence of desmosomal mutations causing the disease and the type of mutation were not associated with specific electrocardiographic, clinical, arrhythmic, anatomic, or prognostic characteristics.
ConclusionsThe incidence of pathological or likely pathological desmosomal mutations in ARVC is very high, with most mutations causing truncation. The presence of desmosomal mutations was not associated with prognosis.
Keywords
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heart condition defined by the progressive replacement of right ventricular myocardium by fibrofatty tissue, which can lead to arrhythmias, sudden cardiac death, and heart failure. As the disease progresses, the left ventricle usually becomes affected, which worsens the prognosis. It is typically inherited in an autosomal dominant manner by mutations in genes encoding desmosomal proteins. Currently, around 40% to 60% of clinically diagnosed patients have at least 1 genetic variant causing the disease.1–8
No prospective randomized trials have examined the use of implantable cardioverter-defibrillators (ICDs) in patients with ARVC, and the independent predictors of arrhythmic events are derived from retrospective studies.9–11 A consensus document developed for the management of this disease divides patients into 3 risk categories: high, intermediate, and low.12 The high-risk category includes patients who have had a cardiac arrest due to sustained ventricular tachycardia or ventricular fibrillation and patients with severe right ventricular dysfunction (fractional area ≤ 17% or ejection fraction ≤ 35%) or severe left ventricular dysfunction (left ventricular ejection fraction ≤ 35%), even without life-threatening ventricular arrhythmias. ICD implantation is recommended in high-risk ARVC patients (class I).12
The aim of the present study was to analyze the incidence of pathogenic or likely pathogenic desmosomal variants in 36 patients with definite high-risk ARVC.
METHODSStudy Design and PopulationThis observational retrospective cohort study included 36 patients with definite high-risk ARVC and an ICD followed up in our hospital between January 2000 and January 2016.
Definite ARVC was diagnosed according to the 2010 modification of the Task Force Criteria,4 namely, at least 2 major criteria, 1 major and 2 minor criteria, or 4 minor criteria from different diagnostic categories.
The high-risk criterion was established according to the consensus document of international cardiology societies,12 namely, patients in secondary prevention due to ventricular tachyarrhythmias or previous cardiac arrest or in primary prevention due to severe right and/or left ventricular systolic dysfunction.
Demographic, clinical, anatomical, electrocardiographic, and arrhythmic data were collected, as well as the number and type of diagnostic criteria met by patients at the time of diagnosis. The demographic characteristics were recorded at disease diagnosis. The anatomical characteristics were obtained using Doppler echocardiography or cardiac magnetic resonance imaging.
The first major arrhythmic event was defined as the first sustained ventricular tachycardia, aborted sudden cardiac death, or appropriate therapy in a patient who had not had any previous event (patients with ICDs in primary prevention).
The study meets the ethical standards of the Declaration of Helsinki, 1975.
GeneticsThe genetic analysis was carried out in the index cases through massively parallel next-generation sequencing between December 2013 and June 2016 and included all of the genes associated with the disease or with other cardiomyopathies. The following genes were studied: DSC2, DSG2, DSP, FLNC, JUP, PKP2, PLN, TMEM43, CTNNA3, DES, LMNA, RYR2, TGFB3, TTN, CASQ2, CTNNB1, LDB3, PERP, PKP4, PPP1R13L, and SCN5A. Bidirectional sequencing of the gene fragment (PKP2 in this case) of the previously identified genetic variant was performed in 1 relative of this index case.
The exons and their intron boundaries were captured using a probe library (SureSelect Target Enrichment Kit; Illumina, Agilent Technologies). The sequencing was performed using the Illumina platform (Illumina HiSeq 1500; San Diego, California, United States) with a read length of 2 × 100 bases according to Illumina protocols. The bioinformatic analysis was carried out through different genotyping programs. Frequency-related information from different populations was considered (1000 Genomes Project, Exome Variant Server, Aggregation Consortium). For variant filtering, an internal strategy of variant filtering and prioritization designed by Health in Code was used, with the analysis focusing on the main genes included in the arrhythmogenic cardiomyopathy panel. A team of cardiologists and geneticists evaluated and classified the variants using the variant classification schema of Health in Code adapted from the American College of Medical Genetics.
The variants considered clinically relevant according to patient phenotype were confirmed by Sanger sequencing.
Follow-upAppropriate events were defined as an ICD intervention involving antitachycardia therapy or as shocks in response to sustained ventricular arrhythmia. Inappropriate events were defined as an ICD intervention with antitachycardia therapy or a shock due to supraventricular tachycardia (atrial fibrillation, atrial or sinus tachycardia) or device malfunction (oversensing or noise).
During follow-up, appropriate ventricular arrhythmic events were recorded (number, time from implantation, cycle length, antitachycardia therapy efficacy, and shocks), as well as inappropriate events (number, time from implantation, and cause), atrial fibrillation or heart failure onset, need for substrate ablation, left ventricular damage, and any deaths or transplants that occurred.
Statistical AnalysisSPSS (version 18.0) was used for the data analysis. Continuous variables are expressed as mean ± standard deviation and categorical variables as absolute values and percentages. The Student t test (for normal distributions) or the Mann-Whitney U test (for nonnormal distributions) was used for the comparison of continuous variables. Categorical variables were compared by means of contingency tables and the chi-square test or Fisher exact test. Arrhythmic event-free time was studied using the Kaplan-Meier method.
Two-sided P values < .05 were considered statistically significant.
RESULTSBaseline Characteristics of the SampleMost patients were male (28 patients, 78%) with a mean age at diagnosis of 45 ± 18 years. All had definite ARVC according to the 2010 modification of the Task Force Criteria,4 independently of the genetic result.
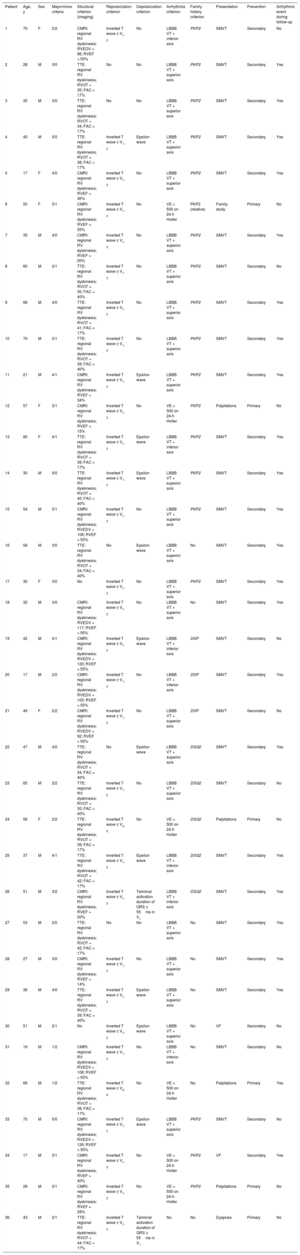
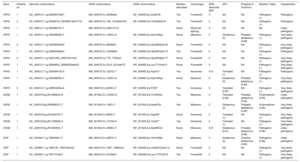
The diagnostic criteria and the main clinical characteristics are shown in Table 1, taking into account that no patients underwent endomyocardial biopsy to establish the diagnosis, although it was confirmed a posteriori in 1 patient because heart transplantat was performed.
Modified Task Force 2010 Criteria and Main Characteristics of the 36 Patients in the Sample
| Patient | Age, y | Sex | Major/minor criteria | Structural criterion (imaging) | Repolarization criterion | Depolarization criterion | Arrhythmia criterion | Family history criterion | Presentation | Prevention | Arrhythmic event during follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 70 | F | 2/2 | CMRI: regional RV dyskinesia; RVEDV = 96; RVEF > 55% | Inverted T wave ≥ V1-3 | No | LBBB VT + inferior axis | PKP2 | SMVT | Secondary | No |
| 2 | 28 | M | 3/0 | TTE: regional RV dyskinesia; RVOT = 35; FAC > 17% | No | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 3 | 35 | M | 3/0 | TTE: regional RV dyskinesia; RVOT = 34; FAC > 17% | No | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 4 | 45 | M | 5/0 | TTE: regional RV dyskinesia; RVOT = 38; FAC > 17% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 5 | 17 | F | 4/0 | CMRI: regional RV dyskinesia; RVEF = 30% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 6 | 50 | F | 3/1 | CMRI: regional RV dyskinesia; RVEF = 35% | Inverted T wave ≥ V1-3 | No | VE > 500 on 24-h Holter | PKP2 (relative) | Family study | Primary | No |
| 7 | 35 | M | 4/0 | CMRI: regional RV dyskinesia; RVEF = 20% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 8 | 60 | M | 3/1 | TTE: regional RV dyskinesia; RVOT = 30; FAC > 40% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | No |
| 9 | 68 | M | 4/0 | TTE: regional RV dyskinesia; RVOT = 41; FAC > 17% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 10 | 70 | M | 3/1 | TTE: regional RV dyskinesia; RVOT = 39; FAC > 40% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 11 | 21 | M | 4/1 | CMRI: regional RV dyskinesia; RVEF = 34% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 12 | 57 | F | 3/1 | CMRI: regional RV dyskinesia; RVEF = 15% | Inverted T wave ≥ V1-3 | No | VE > 500 on 24-h Holter | PKP2 | Palpitations | Primary | No |
| 13 | 80 | F | 4/1 | TTE: regional RV dyskinesia; RVOT = 39; FAC > 17% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + inferior axis | PKP2 | SMVT | Secondary | Yes |
| 14 | 30 | M | 5/0 | TTE: regional RV dyskinesia; RVOT = 40; FAC > 40% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 15 | 54 | M | 3/1 | CMRI: regional RV dyskinesia; RVEDV = 108; RVEF > 55% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 16 | 58 | M | 3/0 | TTE: regional RV dyskinesia; RVOT = 34; FAC > 40% | No | Epsilon wave | LBBB VT + superior axis | No | SMVT | Secondary | Yes |
| 17 | 30 | F | 3/0 | No | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | PKP2 | SMVT | Secondary | Yes |
| 18 | 32 | M | 3/0 | CMRI: regional RV dyskinesia; RVEDV = 117; RVEF > 55% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | No | SMVT | Secondary | Yes |
| 19 | 42 | M | 4/1 | CMRI: regional RV dyskinesia; RVEDV = 120; RVEF > 55% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + inferior axis | DSP | SMVT | Secondary | No |
| 20 | 17 | M | 2/2 | CMRI: regional RV dyskinesia; RVEDV = 105; RVEF > 55% | Inverted T wave ≥ V1-3 | No | LBBB VT + inferior axis | DSP | SMVT | Secondary | Yes |
| 21 | 49 | F | 2/2 | CMRI: regional RV dyskinesia; RVEDV = 92; RVEF > 55% | Inverted T wave ≥ V1-2 | No | LBBB VT + superior axis | DSP | SMVT | Secondary | No |
| 22 | 47 | M | 4/0 | TTE: regional RV dyskinesia; RVOT = 34; FAC > 40% | No | Epsilon wave | LBBB VT + superior axis | DSG2 | SMVT | Secondary | Yes |
| 23 | 65 | M | 2/2 | TTE: regional RV dyskinesia; RVOT = 30; FAC > 40% | Inverted T wave ≥ V1-2 | No | LBBB VT + superior axis | DSG2 | SMVT | Secondary | No |
| 24 | 56 | F | 2/2 | TTE: regional RV dyskinesia; RVOT = 39; FAC > 17% | Inverted T wave ≥ V4-6 | No | VE > 500 on 24-h Holter | DSG2 | Palpitations | Primary | No |
| 25 | 37 | M | 4/1 | TTE: regional RV dyskinesia; RVOT = 42; FAC > 17% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + inferior axis | DSG2 | SMVT | Secondary | Yes |
| 26 | 51 | M | 3/2 | CMRI: regional RV dyskinesia; RVEF = 20% | Inverted T wave ≥ V1-3 | Terminal activation duration of QRS ≥ 55ms in V1 | LBBB VT + inferior axis | DSG2 | SMVT | Secondary | Yes |
| 27 | 53 | M | 2/0 | TTE: regional RV dyskinesia; RVOT = 42; FAC > 17% | No | No | LBBB VT + superior axis | No | SMVT | Secondary | Yes |
| 28 | 27 | M | 3/0 | CMRI: regional RV dyskinesia; RVEF = 14% | Inverted T wave ≥ V1-3 | No | LBBB VT + superior axis | No | SMVT | Secondary | Yes |
| 29 | 36 | M | 4/0 | TTE: regional RV dyskinesia; RVOT = 39; FAC > 40% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + superior axis | No | SMVT | Secondary | Yes |
| 30 | 51 | M | 2/1 | No | Inverted T wave ≥ V1-2 | Epsilon wave | LBBB VT + superior axis | No | VF | Secondary | No |
| 31 | 19 | M | 1/2 | CMRI: regional RV dyskinesia; RVEDV = 108; RVEF > 55% | Inverted T wave ≥ V1-3 | No | LBBB VT + inferior axis | No | SMVT | Secondary | No |
| 32 | 68 | M | 1/2 | TTE: regional RV dyskinesia; RVOT = 38; FAC > 17% | Inverted T wave ≥ V4-6 | No | VE > 500 on 24-h Holter | No | Palpitations | Primary | Yes |
| 33 | 70 | M | 5/0 | CMRI: regional RV dyskinesia; RVEDV = 126; RVEF > 55% | Inverted T wave ≥ V1-3 | Epsilon wave | LBBB VT + superior axis | PKP2 | SMVT | Secondary | No |
| 34 | 17 | M | 3/1 | CMRI: regional RV dyskinesia; RVEF = 40% | Inverted T wave ≥ V1-3 | No | VE > 500 on 24-h Holter | PKP2 | VF | Secondary | Yes |
| 35 | 28 | M | 3/1 | CMRI: regional RV dyskinesia; RVEF = 28% | Inverted T wave ≥ V1-3 | No | VE > 500 on 24-h Holter | PKP2 | Palpitations | Primary | No |
| 36 | 43 | M | 2/1 | TTE: regional RV dyskinesia; RVOT = 44; FAC > 17% | Inverted T wave ≥ V1-3 | Terminal activation duration of QRS ≥ 55ms in V1 | No | No | Dyspnea | Primary | No |
CMRI, cardiac magnetic resonance imaging; DSG2, desmoglein 2; DSP, desmoplakin; F, female; FAC, fractional area change; LBBB VT, ventricular tachycardia with left bundle branch block morphology; M, male; PKP2, plakophilin 2; RV, right ventricle; RVED, right ventricular ejection fraction; RVEDV, RV end-diastolic volume according to body surface area (mL/m2); RVOT, right ventricular outflow tract in the parasternal long-axis projection; SMVT, sustained monomorphic ventricular tachycardia; TTE, transthoracic echocardiography; VE, ventricular extrasystole; VF, ventricular fibrillation.
Regarding disease presentation, most patients were diagnosed on the basis of arrhythmic events: 2 patients had aborted sudden cardiac death and 28 had sustained ventricular tachycardia, with left bundle branch block morphology (20 of the superior axis and 8 of the inferior axis); 4 of these patients were diagnosed after assessment of ventricular extrasystoles, 1 with heart failure during the study and another during the family screening (the single relative of this female index case included in the genetic study, her mother). The first 30 patients were implanted with an ICD for secondary prevention, whereas the remaining 6 received the ICD in primary prevention due to severe ventricular damage (2 biventricular and 4 right ventricular).
The average left ventricular ejection fraction was 58% ± 10%; 6 patients (17%) had left ventricular damage and 2 (6%) had severe ventricular dysfunction (left ventricular ejection fraction < 35%). The average right ventricular ejection fraction was 40% ± 18% on cardiac magnetic resonance imaging, and 18 patients (50%) had severe right ventricular dysfunction (right ventricular ejection fraction < 35%) as measured by echocardiography or magnetic resonance.
Late gadolinium enhancement was found in 10 of the 18 patients who underwent cardiac magnetic resonance imaging: in 7 patients, in the right ventricle; in 2, in both ventricles; and in 1, exclusively in the left ventricle.
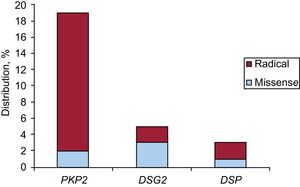
GeneticsNext-generation sequencing detected at least 1 pathogenic or likely pathogenic desmosomal variant in 27 patients (75%): 26 index cases and 1 relative. Of these, plakophilin 2 (PKP2) variants were the most frequent, with 19 variants, followed by 5 desmoglein 2 (DSG2) variants and 3 desmoplakin (DSP) variants.
There were 5 nonsense, 15 frameshift, 1 splice, and 6 missense mutations. Ten novel mutations not previously described were found in 15 patients (71%). One of them (NP_004563.2:p.Glu259Glyfs*77) was detected in 5 index cases of 5 different families in the same province; another (NP_004563.2: p.Gly328Glufs*24) showed cosegregation and, in addition, a family member met the criteria for high-risk disease (right ventricular dysfunction) and was thus included in the genetic study (the single relative of this index case included). Regarding the 8 remaining variants, 49 relatives were studied; 21 were found to be carriers and there was cosegregation in 5 of the variants, but no family member met the criteria for high-risk ARVC.
Figure 1 shows their distribution based on the gene affected and its nature. The characteristics of the variants are detailed in Table 2.

Desmosome Variants of the Study Patients
| Gene | Patients, no. | Genomic nomenclature | cDNA nomenclature | cDNA nomenclature | Mutation described | Variant type | 5000 Genomes MAF | SIFT | Polyphen-2 (HumVar) | Mutation Taster | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PKP2 | 1 | NC_000012.11:g.32945647delT | NM_004572.3:c.2509delA | NP_004563.2:p.Ser837fs | Yes | Frameshift | 0 | NA | NA | Pathogenic (1) | Pathogenic |
| PKP2 | 3 | NC_000012.11:g.33049518_33049521delTCTG | NM_004572.3:c.148_151delACAG | NP_004563.2:p.Thr50Serfs*61 | Yes | Frameshift | 0 | NA | NA | Pathogenic (1) | Pathogenic |
| PKP2 | 1 | NC_000012.11:g.32949237A>T | NM_004572.3:c.2300-5T>A | Novel | Abnormal splicing | 0 | NA | NA | NA | Likely pathogenic | |
| PKP2 | 1 | NC_000012.11:g.33003825G>T | NM_004572.3:c.1253C>A | NP_004563.2:p.Ala418Asp | Novel | Missense | 0 | Deleterious (0) | Probably deleterious (0.99) | Pathogenic (1) | Very likely pathogenic |
| PKP2 | 2 | NC_000012.11:g.33030835delC | NM_004572.3:c.983delG | NP_004563.2:p.Gly328Glufs*24 | Novel | Frameshift | 0 | NA | NA | Pathogenic (1) | Very likely pathogenic |
| PKP2 | 1 | NC_000012.11:g.32994008delC | NM_004572.3:c.1643delG | NP_004563.2:p.Gly548Valfs*15 | Yes | Frameshift | 0 | NA | NA | Pathogenic (1) | Pathogenic |
| PKP2 | 5 | NC_000012.11:g.33031040_33031041insC | NM_004572.3:c.775_776insG | NP_004563.2:p.Glu259Glyfs*77 | Novel | Frameshift | 0 | NA | NA | Pathogenic (1) | Very likely pathogenic |
| PKP2 | 1 | NC_000012.11:g.32949222_32949223delAG | NM_004572.3:c.2312_2313delTC | NP_004563.2:p.Leu771Profs*2 | Novel | Frameshift | 0 | NA | NA | Pathogenic (1) | Very likely pathogenic |
| PKP2 | 1 | NC_000012.11:g.33003841G>A | NM_004572.3:c.1237C>T | NP_004563.2:p.Arg413* | Yes | Nonsense | 0.01 | Tolerated (1) | NA | Pathogenic (1) | Pathogenic |
| PKP2 | 1 | NC_000012.11:g.33003825G>T | NM_004572.3:c.1253C>A | NP_004563.2:p.Ala418Asp | Novel | Missense | 0 | Deleterious (0) | Probably deleterious (0.99) | Pathogenic (1) | Very likely pathogenic |
| PKP2 | 2 | NC_000012.11:g.32955433G>A | NM_004572.3:c.2203C>T | NP_004563.2:p.R735* | Yes | Nonsense | 0 | Tolerated (0.87) | NA | Pathogenic (1) | Very likely pathogenic |
| DSG2 | 1 | NC_000018.9:g.29104840A>G | NM_001943.3:c.1003A>G | NP_001934.2:p.Thr335Ala | Yes | Missense | 0 | Tolerated (0.08) | Probably deleterious (0.94) | Polymorphism (0.52) | Likely pathogenic |
| DSG2 | 1 | NC_000018.9:g.29099820C>T | NM_001943.3:c.136C>T | NP_001934.2:p.Arg46Trp | Yes | Missense | 0 | Deleterious (0) | Probably deleterious (0.99) | Polymorphism (0.99) | Very likely pathogenic |
| DSG2 | 1 | NC_000018.9:g.29102207A>T | NM_001943.3:c.685A>T | NP_001934.2:p.Arg229* | Novel | Nonsense | 0 | Tolerated (1) | NA | Pathogenic (1) | Very likely pathogenic |
| DSG2 | 1 | NC_000018.9:g.29115262G>A | NM_001943.3:c.1310G>A | NP_001934.2:p.Trp437* | Yes | Nonsense | 0 | Tolerated (1) | NA | Pathogenic (1) | Very likely pathogenic |
| DSG2 | 1 | NC_000018.9:g.29126255C>T | NM_001943.3:c.2906C>T | NP_001934.2:p.Ala969Val | Novel | Missense | 0.01 | Deleterious (0.01) | Probably deleterious (0.98) | Pathogenic (0.96) | Likely pathogenic |
| DSP | 1 | NC_000006.11:g.7584082C>T | NM_004415.2:c.6587C>T | NP_004406.2:p.Thr2196Ile | Novel | Missense | 0 | Deleterious (0) | Possibly deleterious (0.90) | Pathogenic (0.96) | Likely pathogenic |
| DSP | 1 | NC_000006.11:g.7585192_7585193insG | NM_004415.2:c.7697_7698insG | NP_004406.2:p.Val2567Cysfs*14 | Novel | Frameshift | 0 | NA | NA | Pathogenic (1) | Very likely pathogenic |
| DSP | 1 | NC_000006.11:g.7581741delT | NM_004415.2:c.5318delT | NP_004406.2:p.Leu1773Tyrfs*8 | Yes | Frameshift | 0 | NA | NA | Pathogenic (1) | Very likely pathogenic |
DSG2, desmoglein 2; DSP, desmoplakin; NA, not applicable; PKP2, plakophilin 2.
In addition, desmosomal variants were classified as probably not pathogenic in 3 patients: 2 in plakoglobin (JUP) (NP_068831.1:p.Lys261Trpfs*39 and NP_068831.1:p.Glu146Lys) and another in desmocollin 2 (DSC2) (abnormal splicing intron 9).
In addition to the desmosomal mutations, other variants of nondesmosomal genes were detected in 4 patients. In particular, there were 3 mutations in the gene encoding titin (TTN) and 1 in the ryanodine receptor gene (RYR2), which has not been associated with any clinical characteristics or specific prognoses. Isolated variants were not detected in non-desmosomal genes associated with the disease.
The PKP2 gene mutation was associated with exclusively right ventricular damage (100% right ventricular damage vs 0% left ventricular or biventricular; P < .05). In the 6 patients with left ventricular damage, 2 had a mutation in DSP (67% vs 33%; P = .07) and 1 had a mutation in DSG2; in the remaining 3, the genetic study was negative.
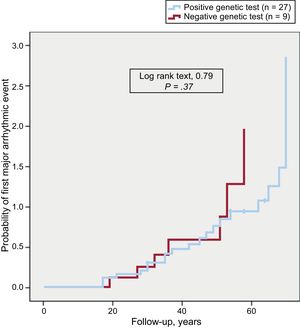
The presence of pathogenic variants, the types of genes affected, or their nature were not associated with an early first arrhythmic event (Figure 2), a distinct clinical presentation, or specific electrocardiographic and anatomical characteristics.
Follow-up
During a median follow-up of 5 [1-20] years, 23 patients (64%) had at least 1 appropriate arrhythmic event according to the ICD. The median time to first event was 7 [1-29] months. A total of 321 ventricular arrhythmias were registered and treated, with a mean cycle length of 283.05 ± 35.68ms; 7 patients were admitted for arrhythmic storm and 5 required substrate ablation for repeated ventricular arrhythmia (3 endocardial and 2 epicardial).
All patients were taking beta-blockers, mainly sotalol (21 patients [58%]), and 11 patients (31%) took amiodarone at some point.
The presence or absence of causal desmosomal variants or their nature was not associated with prognostic features determined by the frequency of ventricular arrhythmia occurrence or their earlier development.
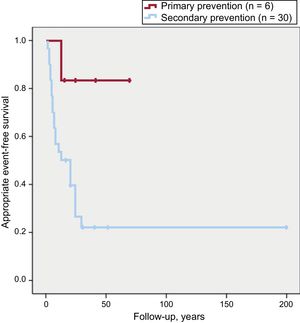
Of the 23 patients who had an appropriate arrhythmic event during follow-up, 22 (96%) had received an ICD for secondary prevention vs 1 for primary prevention (P = .02). The event-free survival according to ICD indication is shown in Figure 3.

Four patients had an inappropriate event during follow-up: 3 due to paroxysmal atrial fibrillation and 1 due to sinus tachycardia; 3 patients had device-related complications: 2 infections and 1 lead fracture.
During follow-up, 7 patients had atrial fibrillation and 3 attended the emergency department due to heart failure. There was 1 heart transplant (for refractory heart failure), but there were no deaths.
DISCUSSIONThe present work details the high incidence of pathogenic or likely pathogenic desmosomal variants associated with high-risk ARVC detected via next-generation sequencing; in addition, a high percentage of these mutations cause truncations that result in the translation of dysfunctional proteins.
The incidence of desmosomal mutations described in previous series varied between 30% and 60%,8 as for example in the series of den Haan et al.,13 where desmosomal variants were detected in 52% of 82 patients with ARVC, and the presence of these mutations was associated with a higher probability of ventricular tachycardia (73% vs 44%) and a younger age at presentation (33 vs 41 years) vs those with negative genetic testing. The Danish series of Christensen et al.14 contained fewer mutations (33% of 65 patients, 10 of them with a borderline diagnosis). The incidence was higher in our series, probably due to good patient selection, because all patients had definite ARVC, most had ventricular arrhythmias, and half had severe damage due to right ventricular contractility. Indeed, a recent article by Medeiros-Domingo et al.15 found a higher percentage of desmosomal mutations in patients with ARVC (n = 7) vs borderline or possible ARVC (n = 7); the latter patients also had changes in other genes more closely related to dilated cardiomyopathy and it was speculated that their conditions would represent phenocopies or an overlap between ARVC and dilated cardiomyopathy.15 These data are consistent with those of the present study, because, after the use of cardiomyopathy gene panels, the mutations detected were desmosomal and only 4 patients had nondesmosomal mutations with questionable pathogenicity: 3 of them in TTN, whose missense mutations tend to have little effect on disease onset and, although they have been associated with ARVC, they are more strongly linked to dilated cardiomyopathy. In addition, their presence was not associated with prognosis or further left ventricular damage. These results may suggest that nondefinite ARVC with negative genetic findings may not be true ARVC but phenocopies or an overlapping clinical presentation with dilated cardiomyopathy. However, more studies are needed to confirm this hypothesis.
New desmosomal variants were found (10 in total) that were considered likely to be pathogenic based on population frequency and bioinformatic prediction data. In addition, cosegregation was detected in 6 of the 9 remaining new variants, although only 1 relative of 1 index case was included in the study (NP_004563.2: p.Gly328Glufs*24) because this individual met the criteria for high-risk disease. The presence of 5 index cases from different families in the same geographical area with the same PKP2 mutation (NP_004563.2: p.Glu259Glyfs*77) indicates the existence of a founder effect, which could be one of the causes of the high rate of positive genetic results in this series. A genetic study is currently being performed in these 5 families to identify a likely common origin.6 In contrast, because 3 variants detected were classified as not associated with the disease, cardiovascular genetics experts must rigorously analyze the results of next-generation sequencing to determine their importance and guide diagnosis and management.16
In the present study, PKP2 mutations were associated with exclusively right ventricular damage; in contrast, as in other studies, DSP mutations17 were associated with damage to the left ventricle (without statistical significance).
In addition, no differences were found in the presence or absence of pathogenic variants or their nature in relation to a larger number of arrhythmic events or their earlier onset. Nonetheless, because this study involves a highly selected group of patients and most (86%) had some arrhythmic event throughout their lifetimes, the identification of differences in prognosis seems complicated. Bhonsale et al.17 published the largest series of patients with ARVC and mutations, and those with radical variants did not have a worse arrhythmic profile, higher mortality, or more transplants, although the profile of these patients was different from that of ours, with only 60% having definite ARVC, 38% having ventricular arrhythmias, and 44% having an ICD.
The importance of genetic testing in this population is due to the confirmation of the diagnosis (Task Force diagnostic criteria4) and, in the family study, due to a possible early detection of the disease, the recommendation to avoid competitive sports, and the ability to avoid repeated tests and the stress caused by long-term follow-up of patients with negative genetic findings.
The use of ICD devices in this high-risk group is clearly justified (class I indication in the clinical practice guidelines12); most patients (64%) showed appropriate arrhythmic events but, nonetheless, there was no total or cardiovascular mortality during follow-up and only 1 patient required transplant (for refractory heart failure).
Notably, ICD recipients in secondary prevention had worse arrhythmic event-free survival than those in primary prevention. Only 1 of the 6 patients in primary prevention had a treated ventricular arrhythmia, which highlights the difficult selection of these patients, particularly because the risk factors associated with a worse arrhythmic prognosis are based on retrospective, nonrandomized studies, most of which included patients who already had a major arrhythmic event that motivated the ICD implantation.12
LimitationsThe present work comprises a retrospective and single-center study, with a small number of patients with high-risk ARVC, and the data were not compared with those of moderate- or low-risk disease. In addition, the study did not include post mortem studies that would most probably have revealed a high arrhythmic risk.
Given the retrospective nature of the study, confounding factors might have affected the results.
CONCLUSIONSThe incidence of pathogenic or likely pathogenic desmosomal variants in definite high-risk ARVC patients is very high and most mutations are radical. The presence of these mutations or their more unstructured nature was not associated with specific prognostic characteristics.
Despite having the same degree of recommendation in international consensus guidelines, arrhythmic event-free survival is significantly better in patients with high-risk ARVC in primary prevention than in those in secondary prevention.
CONFLICTS OF INTERESTNone declared.
- –
Arrhythmogenic right ventricular cardiomyopathy is a heart disease defined as the progressive replacement of right ventricular myocardium by fibrofatty tissue that can lead to arrhythmias, sudden cardiac death, and heart failure. Currently, between 40% and 60% of patients have at least 1 genetic mutation associated with the disease.
- –
The present next-generation sequencing study detected a high incidence of mutations in desmosomal genes associated with definite ARVC according to the Task Force diagnostic criteria and of high-risk disease according to the consensus document of international societies; most mutations caused truncations.
The authors appreciate the technical support enabling this work provided by the Health in Code laboratory of cardiovascular genetics.