Hereditary pulmonary veno-occlusive disease (PVOD) has been associated with biallelic mutations in EIF2AK4 with the recent discovery of a founder mutation in Iberian Romani patients with familial PVOD. The aims of this study were phenotypical characterization and survival analysis of Iberian Romani patients with familial PVOD carrying the founder p.Pro1115Leu mutation in EIF2AK4, according to their tolerance to pulmonary vasodilators (PVD). Familial genetic screening was conducted, as well as assessment of sociocultural determinants with a potential influence on disease course.
MethodsObservational study of Romani patients with familial PVOD included in the Spanish Registry of Pulmonary Arterial Hypertension. Genetic screening of EIF2AK4 was performed in index cases and relatives between November 2011 and July 2016 and histological pulmonary examination was carried out in patients who received a lung transplant or died. The patients were divided into 2 groups depending on their tolerance to PVD, with comparison of baseline characteristics and survival free of death or lung transplant.
ResultsEighteen Romani patients were included: 9 index cases and 9 relatives. The biallelic founder mutation in EIF2AK4 was found in all affected cases and 2 unaffected relatives. Family screening showed 34.2% of healthy heterozygotes, high consanguinity, young age at childbirth, and frequent multiparity. Prognosis was bleak, with significant differences depending on tolerance to PVD.
ConclusionsWe describe 2 phenotypes of hereditary PVOD depending on tolerance to PVD, with prognostic impact and familial distribution. Consanguinity may have a negative impact on the transmission of PVOD, with familial genetic screening showing high effectiveness.
Keywords
Pulmonary veno-occlusive disease (PVOD) is a rare, aggressive form of pulmonary arterial hypertension (PAH) characterized by pulmonary venous involvement with a progressive increase in pulmonary vascular resistance, hypoxemic respiratory failure, right ventricular failure, and eventual death.1–4
Diagnosis is based on the integration of compatible clinical and radiological findings and observation of a marked decrease in diffusing capacity of the lung for carbon monoxide (DLCO) and suggestive bronchoalveolar lavage findings. Histologic confirmation is necessary for a definitive diagnosis.5 Clinical presentation is diverse and mutations in the EIF2AK4 gene have been described in familial and sporadic forms of PVOD/pulmonary capillary hemangiomatosis.2,6,7 No other genes have been implicated to date and detection of a homozygous EIF2AK4 mutation is considered diagnostic of PVOD/pulmonary capillary hemangiomatosis.8 A homozygous founder mutation in the EIF2AK4 gene—c.3344C>T(p.Pro1115Leu)—was described in 18 Romani patients with an aggressive form of familial PVOD characterized by an early age at diagnosis, severely reduced DLCO, and short survival.8–10
The evidence on tolerance of pulmonary vasodilators in PVOD is contradictory. There are no predictors of a favorable response1,2 and this form of treatment is not recommended in this setting, leaving lung transplantation as the only effective option.11,12
The main aims of this study were to perform phenotypic characterization and analyze survival free of death or lung transplantation in a cohort of Iberian Romani patients with familial PVOD carrying the founder EIF2AK4 mutation (p.Pro1115Leu) according to clinical tolerance of treatment with pulmonary vasodilators. Secondary aims were a) to estimate the prevalence of heterozygous and homozygous carriers through genetic screening, and b) to analyze sociocultural aspects associated with the Romani population that might influence the transmission of PVOD.
METHODSThis study formed part of the Spanish multicenter pulmonary hypertension genetic project. We conducted an observational study of Romani patients with familial PVOD from the Spanish Pulmonary Hypertension Registry (REHAP Registry). The genetic study was performed between November 2011 and July 2016. A tentative diagnosis of PVOD was established on clinical grounds (precapillary PAH and reduced DLCO) and a compatible computed tomography pattern (mediastinal lymph node enlargement, septal thickening, and centrilobular nodules). Patients were diagnosed with familial PVOD if they had 1 or more relatives with a confirmed diagnosis of PVOD or relatives (alive or deceased) with a history suggestive of PVOD. The clinical data from the REHAP Registry were included in the analysis13 ().
Patients were classified into 2 subgroups according to their long-term tolerance of treatment with pulmonary vasodilators: a) nontolerant patients, ie, those who developed clinical or subclinical pulmonary edema or experienced worsening of respiratory failure (increased desaturation at rest or on exertion) following treatment initiation, and b) tolerant patients, ie, those who experienced clinical improvement (improved functional class) or improved hemodynamic parameters (reduction in pulmonary vascular resistance and/or right atrial pressure and/or an increase in cardiac index) in consecutive visits following treatment initiation.
Genetic StudyPeripheral blood was collected from probands and relatives following provision of informed consent. The genetic study was conducted at the Institute of Medical and Molecular Genetics at Hospital Universitario La Paz in Madrid, Spain ().
Statistical AnalysisThe statistical analysis was performed using IBM SPSS Statistics, v.22 (SPSS, Chicago, Illinois, USA). Data are expressed as mean ± SD, and statistical significance was set at P ≤ .05.
Survival analysis was conducted with Kaplan-Meier curves using the date of diagnosis as the start date and completion of follow-up (October 15, 2016) as the end date. The event of interest was death or lung transplantation.
The log-rank test was used to compare survival between PVOD patients carrying the EIF2AK4 mutation according to whether or not they tolerated treatment with pulmonary vasodilators.
Family Study and Sociocultural ProfileA genetic study was offered to all first- and second-degree relatives, the spouses of heterozygous and homozygous carriers, and their children.
At the first genetic counseling visit, a pedigree chart was drawn up and samples were collected from probands. The results of the genetic study were given at the second visit.
Information was gathered on consanguinity, number of pregnancies per patient, and age at first pregnancy.
Histopathologic StudyHistopathologic pulmonary changes were evaluated in deceased patients who had agreed to an autopsy and in patients who underwent lung transplantation, as per the protocol described in .
The study was conducted in compliance with the principles of the Declaration of Helsinki and the Spanish Law for the Protection of Personal Data and was approved by the ethics committees at the participating hospitals. All patients gave their written informed consent.
RESULTSBetween November 1, 2011 and July 1, 2016, 18 Romani patients with familial PVOD from the REHAP Registry (9 probands and 9 relatives) underwent genetic testing. As of July 1, 2016, the REHAP Registry contained data on 78 patients with PVOD; 19 (24.3%) had familial PVOD and 59 had another form of this disease. Of the 19 patients with familial PVOD, 18 were of Romani ethnicity and were included in our study.
We identified 9 probands, all carriers of the founder EIF2AK4 mutation c.3344C>T(p.Pro1115Leu). They belonged to 9 families with a distinct ethnic origin (8 from northern Spain and 1 from Portugal). The other 9 patients were first-degree relatives of the probands. They had been diagnosed with the disease through family screening and were therefore included in the family study.
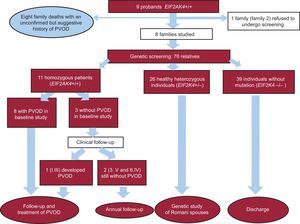
Family StudyMutation screening was offered to all 9 families. One family refused to undergo screening and another agreed at a later date and was awaiting the results at the time of writing this article. We therefore studied 7 families consisting of 76 relatives in total. Thirty-seven of the family members were carriers of the mutation (11 homozygous and 26 heterozygous) and 39 were noncarriers (Fig. 1). Of the 11 homozygous carriers, 8 had diagnostic criteria of PVOD at the initial evaluation (mean ± SD age, 30 ± 8.4 years; 50% male) and 1 developed the disease later (at the age of 17 years).

The family study thus identified 18 cases of familial PVOD (9 probands and 9 relatives). Examination of the family history identified an additional 8 relatives who had died at an early age with a history suggestive of PVOD, although they did not have a confirmed diagnosis and had not undergone genetic testing (Fig. 1). The pedigree charts are shown in .
Mutation ScreeningThe genetic study revealed a missense variant at exon 23 of the EIF2AK4 gene c.3344C>T(p.Pro1115Leu), which was homozygous in the 9 probands. The mutation was considered pathogenic due to the high conservation of Pro1115 throughout evolution, the bioinformatic prediction results, the low frequency of the mutation in the general population, and its cosegregation with the disease ().
The estimated penetrance was 90% (18/20), and at the time of the study, there were 2 healthy homozygous carriers. No phenotypes were detected in the heterozygous carriers.
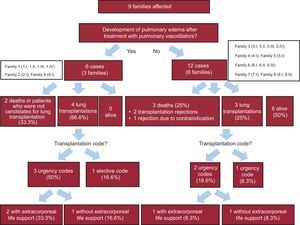
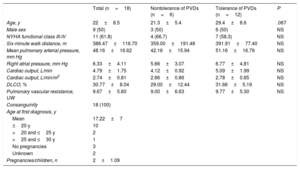
Description of Affected PatientsWe studied the baseline clinical characteristics (Table 1) of the 18 homozygous patients and the progression of their disease. On the basis of their response to pulmonary vasodilators, 6 patients from 3 families were classified as nontolerant and 12 patients from 6 families were classified as tolerant (Fig. 2 and Fig. 3). No differences were observed for tolerance within families, ie, all the members of a given family either tolerated the treatment or did not.
Baseline Characteristics of Homozygous Patients With Pulmonary Veno-occlusive Disease
| Total (n=18) | Nontolerance of PVDs (n=6) | Tolerance of PVDs (n=12) | P | |
|---|---|---|---|---|
| Age, y | 22±8.5 | 21.3±5.4 | 29.4±8.6 | .067 |
| Male sex | 9 (50) | 3 (50) | 6 (50) | NS |
| NYHA functional class III-IV | 11 (61.8) | 4 (66.7) | 7 (58.3) | NS |
| Six-minute walk distance, m | 386.47±116.70 | 359.00±191.48 | 391.91±77.40 | NS |
| Mean pulmonary arterial pressure, mm Hg | 48.16±16.62 | 42.16±15.94 | 51.16±16.79 | NS |
| Right atrial pressure, mm Hg | 6.33±4.11 | 5.66±3.07 | 6.77±4.81 | NS |
| Cardiac output, L/min | 4.79±1.75 | 4.12±0.92 | 5.09±1.99 | NS |
| Cardiac output, L/min/m2 | 2.74±0.81 | 2.66±0.86 | 2.78±0.85 | NS |
| DLCO, % | 30.77±8.04 | 29.00±12.44 | 31.66±5.19 | NS |
| Pulmonary vascular resistance, UW | 9.67±5.60 | 9.00±6.63 | 9.77±5.30 | NS |
| Consanguinity | 18 (100) | |||
| Age at first diagnosis, y | ||||
| Mean | 17.22±7 | |||
| ≤20 y | 10 | |||
| >20 and ≤25 y | 2 | |||
| >25 and ≤30 y | 1 | |||
| No pregnancies | 3 | |||
| Unknown | 2 | |||
| Pregnancies/children, n | 2±1.09 | |||
DLCO, diffusing capacity for carbon monoxide; NS, nonsignificant; NYHA; New York Heart Association; PVD, pulmonary vasodilators.
Values are expressed as No. (%) or mean ± SD unless otherwise specified.
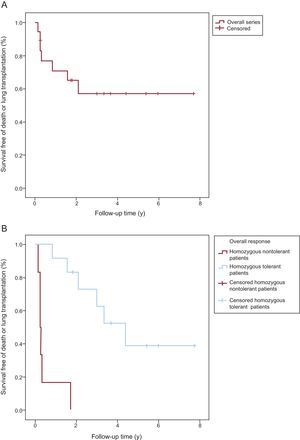
. B: Homozygous patients with PVOD who tolerated specific treatment with pulmonary vasodilators vs those who did not.")
Death-free or lung transplantation–free Kaplan-Meier survival curves. Comparison of survival. A: Overall series of homozygous patients with pulmonary veno-occlusive disease (PVOD). B: Homozygous patients with PVOD who tolerated specific treatment with pulmonary vasodilators vs those who did not.

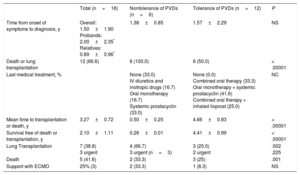
Table 2 and Figure 3 summarize the clinical course and survival for the 18 homozygous carriers and for the subgroups of tolerant and nontolerant patients. Heterozygous patients and asymptomatic homozygous patients were not included.
Summary of Disease Course: Treatment Received, Adverse Events, and Survival of Homozygous Patients With Pulmonary Veno-occlusive Disease
| Total (n=18) | Nontolerance of PVDs (n=6) | Tolerance of PVDs (n=12) | P | |
|---|---|---|---|---|
| Time from onset of symptoms to diagnosis, y | Overall: 1.50±1.90 Probands: 2.00±2.35* Relatives: 0.89±0.96* | 1.38±0.85 | 1.57±2.29 | NS |
| Death or lung transplantation | 12 (66.6) | 6 (100.0) | 6 (50.0) | < .00001 |
| Last medical treatment, % | None (33.0) IV diuretics and inotropic drugs (16.7) Oral monotherapy (16.7) Systemic prostacyclin (33.0) | None (0.0) Combined oral therapy (33.3) Oral monotherapy + systemic prostacyclin (41.6) Combined oral therapy + inhaled iloprost (25.0) | NC | |
| Mean time to transplantation or death, y | 3.27±0.72 | 0.50±0.25 | 4.66±0.83 | < .00001 |
| Survival free of death or transplantation, y | 2.10±1.11 | 0.26±0.01 | 4.41±0.99 | < .00001 |
| Lung Transplantation | 7 (38.8) 3 urgent | 4 (66.7) 3 urgent (n=3) | 3 (25.0) 2 urgent | .002 .225 |
| Death | 5 (41.6) | 2 (33.3) | 3 (25) | .001 |
| Support with ECMO | 25% (3) | 2 (33.3) | 1 (8.3) | NS |
ECMO, extracorporeal membrane oxygenation; IV, intravenous; NC, not calculated; NS, nonsignificant; PVDs, pulmonary vasodilators.
Unless otherwise specified, values are expressed as NoD. (%) or mean±standard deviation.
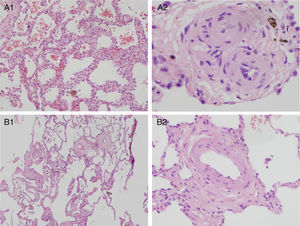
Histopathologic pulmonary changes were examined in 8 patients (by autopsy in 2 cases and by pneumonectomy after lung transplantation in 6). This study was not performed in the remaining 10 patients (4 deceased and 6 alive). The results identified 2 distinct, family-specific profiles. The histopathologic changes observed in families 1, 2, and 9 were characteristic of PVOD. The changes in family 3 were less severe and were consistent with PVOD at a less advanced stage (). Representative images for the 2 subgroups are shown in Figure 4.
. A1: Magnification 4×. Lung parenchyma with septal thickening and evident capillary hemangiomatosis. Intra-alveolar hemosiderin-laden macrophages. A2: Magnification 40×. Fibrotic, thickened veins. B: Histopathologic pulmonary findings in a patient who responded to vasodilators (patient 3.l). B1: Magnification 4×. Less pronounced, diffuse septal thickening, capillary hemangiomatosis, and intraalveolar hemosiderin-laden macrophages. Thickening of vascular wall structures. B2: Magnification 20×. Vascular structure of thickened wall. Note the normal thickness of the alveolar septum at the edge of the image.")
Histopathologic pulmonary findings in homozygous patients with pulmonary vaso-occlusive disease. Hematoxylin-eosin. A: Histopathologic pulmonary findings in patients who did not tolerate vasodilators (patient 1.l). A1: Magnification 4×. Lung parenchyma with septal thickening and evident capillary hemangiomatosis. Intra-alveolar hemosiderin-laden macrophages. A2: Magnification 40×. Fibrotic, thickened veins. B: Histopathologic pulmonary findings in a patient who responded to vasodilators (patient 3.l). B1: Magnification 4×. Less pronounced, diffuse septal thickening, capillary hemangiomatosis, and intraalveolar hemosiderin-laden macrophages. Thickening of vascular wall structures. B2: Magnification 20×. Vascular structure of thickened wall. Note the normal thickness of the alveolar septum at the edge of the image.
The sociocultural variables studied are shown in Table 1. In brief, we observed a 100% consanguinity rate, multiple pregnancies (2.0±1.1 children per patient), and pregnancy at an early age (mean age at first pregnancy of 17.2±7 years).
DISCUSSIONOur group previously described a homozygous founder mutation in the EIF2AK4 gene in 18 patients from 9 Iberian Romani families with aggressive forms of familial PVOD; the disease was transmitted in an autosomal recessive manner and had a penetrance of close to 100%.9,10 Although all the patients studied had severe forms of PVOD, we identified 2 previously undescribed clinical phenotypes, with prognostic implications, based on tolerance of pulmonary vasodilators and histopathologic pulmonary findings. It is particularly interesting that the phenotypes were family-specific.
The REHAP Registry currently features 78 cases of hereditary and idiopathic PVOD. Our study is the first to describe the natural history of Romani patients with familial PVOD carrying a founder EIF2AK4 mutation and to analyze the potential impact of sociocultural practices on disease course and transmission in this population. We studied 23% of all PVOD cases registered in Spain and all cases of hereditary PVOD but one (that of a Caucasian patient). Biallelic EIF2AK4 mutations were detected in all the patients studied, supporting previous findings.2
EIF2AK4 encodes a serine/threonine kinase that regulates the synthesis of proteins in response to cellular stress and amino acid deprivation. However, the mechanisms by which loss of function of this gene leads to the development of PVOD are unknown. Although more studies are needed, it would seem likely that the founder mutation in this population leads to a loss of function of the protein, which, considering the transcript position, might affect its ability to respond to protein deprivation.
Clinical Phenotype and PrognosisAs mentioned, our group previously reported 18 cases of aggressive familial PVOD in Romani patients with a founder mutation in the EIF2AK4 gene.9,10 Montani et al.2 described an additional 26 cases of familial PVOD in 13 families in which they detected 18 EIF2AK4 mutations. The majority were premature stop codons or small deletions or insertions2,7,14 and were associated with marked phenotypic heterogeneity, which is consistent with the underlying genetic heterogeneity. We have described the clinical phenotype of hereditary PVOD in carriers of a founder EIF2AK4 mutation. Although previous studies have reported founder mutations in Romani people with different diseases,15 our group was the first to describe such a mutation in association with PAH.9,10
We observed considerable phenotypic heterogeneity among the families in our series that cannot be attributed to the existence of different genetic bases. In addition, we have described for the first time 2 clinical phenotypes that were clearly distinct in terms of tolerance of pulmonary vasodilators, aggressiveness, and histopathologic findings. These phenotypes may ultimately prove useful for prognostic stratification and treatment. There were 2 extreme cases represented by 2 families (1 and 3). Both families had 4 members who developed severe, early-onset PVOD, but they differed considerably in terms of disease progression and response to pulmonary vasodilators. The disease followed a highly aggressive course in family 1, with a mean ± SD survival of 0.64±0.72 years. Three of the members (75%) required lung transplantation and the fourth (25%) died. Tolerance of pulmonary vasodilators was very poor and the treatment had to be withdrawn in 3 patients. This phenotype contrasts with that observed in family 3, where all 4 members tolerated the use of these drugs. Mean survival was also longer in this family (2.28±0.93 years) and there were 2 cases of lung transplantation (50%) and no deaths. The differences in outcomes between the families were not attributable to differences in baseline DLCO, which was comparable in both families and lower than the rate of 34% described by a French group for a series of patients with PVOD.4
DLCO has been identified as a marker of poor prognosis in PAH. However, in our series, we cannot confirm a relationship between DLCO and PVOD. DLCO was not a predictor of outcome and the reduction in diffusing capacity was similar in tolerant and nontolerant patients, although the second group had more severe disease and a worse prognosis.
There were, however, significant differences in the histopathologic changes observed. The first family (family 1) had considerable venous involvement and foci consistent with pulmonary capillary hemangiomatosis, reflecting an advanced stage of disease, while the second family (family 3) had much less severe changes.
Our hypothesis is that the differences observed in disease course between the 2 families, and, by extension, between those who tolerated pulmonary vasodilator treatment and those who did not, could be partly due to the fact that a higher proportion of responders received specific treatment. However, these differences could also be due to the considerable disparity in disease severity reflected by the histopathologic changes. It should be noted, however, that despite the differences between the 2 patient subgroups, overall prognosis was dismal, with a median survival of close to 2 years and an overall death or lung transplantation rate of 66.6%, which is lower than the rate of 82.2% described by the French group.2,4 It is also noteworthy that survival was worse in our series than in the French series, even though a higher proportion of patients (10/18) were diagnosed when their symptoms were less severe (in the family study). Their baseline profile was also similar,7 namely, severe precapillary PAH, a male to female ratio of 1,3,4 (absent in sporadic PVOD, PVOD associated with exposure to toxic agents, and idiopathic PAH)16,17; similar ages of presentation (27±8 years vs 27±10 years), and slightly more deteriorated functional status (61.8% vs 80% of patients with functional class III/IV). In addition, we identified at least 8 relatives who had died with a history suggestive of PVOD.
The above observations indicate that our patients had a particularly aggressive variant of PVOD, characterized by a phenotypic spectrum in which worse tolerance of pulmonary vasodilators was associated with greater histologic involvement and a worse prognosis. Considering that all the patients in our series carried the same mutation, future studies should investigate additional factors that might influence disease severity, such as genetic, epigenetic, and environmental factors.
Family StudyThe patients in our series belonged to large, consanguineous families in which several members had PVOD. Montani et al.2 reported a lower rate of consanguinity (30%) and fewer affected family members.
Our findings show a high rate of heterozygous carriers (34.2%) and a high diagnostic yield for family screening, which detected early disease in 8 homozygous patients and presymptomatic disease in 3 initially healthy homozygous patients. One of these (a nonresponder to pulmonary vasodilators) developed PVOD 6 months after the genetic study, and the other 2, both responders, are still healthy at the ages of 34 and 38 years old. We cannot, however, rule out the possibility of incomplete penetrance at this age, as there are cases of patients aged 38 years old who developed PVOD despite tolerance of pulmonary vasodilator treatment.
Study PopulationThe Romani are an ethnic group with strong traditions that have an impact on health status and also possibly on the course and transmission of PVOD. Few studies have investigated the Romani population in Spain, despite their considerable size (approximately 750 000 individuals)18 and the fact that they constitute a health risk group. There is evidence that the Romani population have a worse health situation than the general population, despite their younger age,19 and this is probably due to unfavorable social conditions.18
Our study is the first to analyze the influence of sociocultural aspects of Romani lifestyle on the transmission of PVOD. We found several key aspects that exerted a negative effect. First, patients were diagnosed only when their disease was already at an advanced stage. This can be explained by the diagnostic challenges associated with PVOD and the lack of familiarity with the condition among both health professionals and the general population. The delay between symptom onset and diagnosis was similar to that reported for PAH in Spain (mean, 2.2 years).13 The rapid disease progression (with marked deterioration over the course of a few months) also probably contributed to this delay in diagnosis, highlighting the need for more studies on the natural history of PVOD.
Other sociocultural factors that may have had a negative impact on disease outcome include unfavorable social conditions18 and certain traditions related to reproduction and family planning, such as a rejection of contraception, pregnancy at an early age, and multiple pregnancies.18 Age at first pregnancy was significantly lower than that described for the Spanish population (17.2±7 years vs 31.2 years20), and the number of children per mother was also higher (2.01±1.09 vs 1.2721). In addition, although the patients in our series were seriously ill from a young age (22±8.6 years), all their pregnancies occurred before they were 30 years of age.
Considering the recessive mode of inheritance of the EIF2AK4 mutation, the high frequency of endogamy in the Romani population is likely to have an important impact on the transmission of PVOD, as it would favor the appearance of new homozygous carriers. As this mutation, which is autosomal recessive and associated with death at a young age, has only been described to date in the Romani population, it would probably have become extinct had it not emerged in a population with sociocultural characteristics that strongly favor self-perpetuation and propagation.
Considering the findings of the present study, we believe that it is essential that clinicians be alert to the possibility of PVOD when evaluating Romani patients with dyspnea. PVOD should be suspected in patients with a family history suggestive of PVOD, reduced DLCO, and characteristic radiologic findings,2,8 and genetic testing for EIF2AK4 mutations should be ordered on confirmation of PAH. Once a diagnosis of hereditary PVOD has been established, patients should be referred for genetic counseling and a family study conducted. These measures, together with discussion of family planning options, will help to prevent new cases.
LimitationsThis study has several limitations. First, we have only studied a proportion of the Romani families in the REHAP Registry and not all of them have been studied in depth, thereby limiting the conclusions that can be drawn in relation to phenotypic characterization and prognosis. In addition, because we studied Romani patients from consanguineous families, our findings may not be applicable to other populations with hereditary PVOD. Furthermore, the proportion of homozygous and heterozygous patients identified may have been affected by selection bias and may not be representative of other populations. Finally, we distinguished between 2 clinical and histologic phenotypes, but we did not conduct a genetic or epigenetic study to find a possible explanation for the differences observed. More studies are needed to expand our understanding of the pathophysiological and epidemiological aspects of hereditary PVOD.
CONCLUSIONSWe have provided a detailed description of the first series of patients with familial PVOD carrying a founder mutation in the EIF2AK4 gene that appears to be specific to the Iberian Romani population and is associated with a rare, aggressive form of the disease.
Considering the high rate of heterozygous carriers detected and their relevance, family screening has a high yield in this setting, due to its potential role in preventing new cases, as the sociocultural traditions of the Romani population may have favored the transmission and unfavorable progression of this disease in this setting. Finally, we have described 2 new clinical patterns of hereditary PVOD that were family-specific and had prognostic implications.
FUNDINGThis project was partially funded by the Cardiovascular Research Network of Instituto de Salud Carlos III de Madrid (RD06/0003/0012) project FIS 15/02012, and by an unconditional grant from the Spanish National Association of Pulmonary Hypertension, Actelion, and the Air Liquide Foundation.
CONFLICTS OF INTERESTP. Escribano Subías has received unconditional educational grants from Actelion and the Air Liquide Foundation for the current research project; served as a consultant for Actelion, Bayer, Pfizer, and GSK; received speaker's fees from Actelion, Bayer, Pfizer, and GSK; and fees for educational presentations from GSK.
H. Bueno Zamora has received fees for consultancy work, talks, and grants for attending conferences from Abbott, Astra-Zeneca, Bayer, BMS, Ferrer, MEDSCAPE-the heart.org, Novartis, and Servier and fees for research projects from Astra-Zeneca, BMS, Janssen, and Novartis. P. Navas Tejedor has received unconditional educational grants from Actelion and the Air Liquide Foundation.
- –
PVOD is a rare form of PAH that responds poorly to treatment and has a dismal prognosis. Recessive mutations in the EIF2AK4 gene have been associated with hereditary forms of PVOD and a founder mutation has been described in Iberian Romani patients with an aggressive form of hereditary PVOD.
- –
We describe, for the first time, the clinical phenotype and disease course of PVOD in Romani patients carrying a founder EIF2AK4 mutation and report on 2 distinct clinical and histopathologic phenotypes with prognostic implications. We also analyze certain sociocultural aspects of the Romani lifestyle that might influence the transmission and progression of PVOD.
.
We thank the NHLBI GO Exome Sequencing Project and Exome Aggregation Consortium for their enormous contribution to science. We also thank all the patients and families who made this study possible.