Palabras clave
INTRODUCCION
La elevación sostenida de la presión arterial (PA) se asocia con un incremento significativo de la morbimortalidad cardiovascular en los pacientes hipertensos. Ello es debido a que la hipertensión arterial (HTA) puede dañar la estructura y alterar la función de las arterias, el corazón, el cerebro y el riñón. Concretamente, los pacientes con HTA están expuestos a desarrollar una serie de alteraciones estructurales y funcionales del corazón que constituyen la llamada cardiopatía hipertensiva (CH). La hipertrofia ventricular izquierda (HVI) constituye la lesión macroscópica propia de la CH, pero en esta lesión subyacen una serie de cambios microscópicos que configuran un cuadro denominado remodelado miocárdico. La hipertrofia y la apoptosis de los cardiomiocitos, la fibrosis del miocardio y la hipertrofia de la pared de las arterias y las arteriolas intramiocárdicas constituyen los elementos estructurales definitorios del remodelado miocárdico presente en la CH.
BASES MOLECULARES Y CELULARES
Como otros órganos, el corazón está compuesto por células parenquimatosas altamente diferenciadas, los cardiomiocitos, y por estroma constitutido por la matriz extracelular, el fluido tisular y células mesenquimales indiferenciadas pluripotenciales. La matriz extracelular cardiaca está integrada principalmente por colágeno fibrilar y no fibrilar, así como por fibras de laminina y elastina, proteoglicanos e integrinas. Las moléculas de colágeno fibrilar de tipo I y de tipo III son las más abundantes en el corazón adulto y exhiben su típica conformación de triple hélice merced a la disposición espacial de sus 3 cadenas polipeptídicas α. El colágeno fibrilar sirve de andamiaje estructural para los cardiomiocitos y la vasculatura intramiocárdica, a la par que confiere al tejido miocárdico la rigidez que lo hace resistente a la deformación durante el ciclo cardiaco. Además, el colágeno fibrilar conecta los elementos contráctiles de los cardiomiocitos adyacentes, actuando así como un transductor de la contracción del músculo cardiaco hacia la cámara ventricular. Aunque en determinadas situaciones patológicas del corazón hay un déficit de colágeno fibrilar, la mayoría de las cardiopatías crónicas se caracterizan por la acumulación exagerada focal (cicatriz) o difusa (fibrosis) de éste1.
Estudios realizados en corazones humanos post mortem2 y en biopsias endomiocárdicas3 muestran que la fracción de volumen de colágeno miocárdica (FVC, una medida de la cantidad de fibras de colágeno que están depositadas en el miocardio) es significativamente mayor en pacientes con CH que en sujetos normotensos de control. Desde el punto de vista histológico, la fibrosis miocárdica hipertensiva presenta las siguientes características definitorias4-6: está constituida por el depósito exagerado de fibras de colágeno de tipo III inicialmente, y de tipo I a medida que el proceso progresa; las fibras se disponen como haces que surcan el intersticio y en torno a los vasos intramiocárdicos; la acumulación de fibras no se limita al ventrículo izquierdo y también está presente en las otras cámaras cardiacas; y la cuantía del depósito de fibras se relaciona inversamente con el número de cardiomiocitos y directamente con el grado de hipertrofia de éstos.
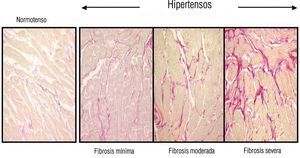
La prevalencia de la fibrosis miocárdica en la CH está poco estudiada. En un estudio en el que se establecían grados de fibrosis a partir de la comparación de los valores de la FVC entre sujetos normotensos sin HVI y pacientes hipertensos con HVI se comprobó que el 11% de los pacientes presentaba una fibrosis nula-mínima, el 58% presentaba una fibrosis ligera-moderada y el 31% presentaba fibrosis severa (fig. 1)3. Por lo tanto, la fibrosis es una lesión prácticamente constante en el miocardio de los pacientes con CH.

Fig. 1. Contenido de colágeno presente en el corazón de un sujeto normotenso y de 3 pacientes con cardiopatía hipertensiva. Los pacientes están clasificados según el grado de fibrosis miocárdica: mínima (izquierda), moderada (centro) y severa (derecha). Las secciones están teñidas con rojo picrosirio y las fibras de colágeno aparecen teñidas de color rojo.
MECANISMOS CAUSALES
El exceso de fibras de colágeno miocárdico presente en la CH es el resultado de la combinación de un incremento en la síntesis de colágeno, por los fibroblastos y los miofibroblastos, y una disminución o ausencia de cambio en la degradación de éste, por las metaloproteinasas de matriz7. Esta hipótesis se apoya en hallazgos experimentales que muestran una sobreexpresión de los genes8 del procolágeno de tipo I (precursor del colágeno de tipo I)8 y una disminución de la actividad de la colagenasa (enzima encargada de la degradación del colágeno de tipo I)9 en el ventrículo izquierdo hipertrofiado de las ratas espontáneamente hipertensas (SHR). La combinación de distintos factores (hemodinámicos, humorales, genéticos y ambientales) puede inducir el citado desequilibrio.
Factores hemodinámicos
Experimentos in vivo han demostrado que la sobrecarga crónica de presión estimula tanto la expresión de genes como la síntesis de proteínas de colágeno en el miocardio, lo que favorece un exagerado depósito de fibras de colágeno y la consiguiente fibrosis10. Además, estudios in vitro han demostrado que la síntesis de procolágeno de tipo I está estimulada en los fibroblastos cardiacos con una sobrecarga mecánica cíclica10, como la que se produce en condiciones de HTA. Por tanto, la sobrecarga hemodinámica del ventrículo izquierdo debida a HTA puede favorecer la fibrosis miocárdica.
Varias observaciones clínicas sustentan esta posibilidad. Tanaka et al11 objetivaron que la FVC aumentaba desde el exterior hasta el interior de la pared libre del ventrículo izquierdo en corazones humanos hipertensos, lo que probablemente refleja el gradiente transmural del estrés parietal. Rossi2 describió que cuando los corazones de pacientes hipertensos se agrupaban según el peso del corazón, la FVC aumentaba progresivamente con el peso cardiaco. Además, había una asociación entre la severidad de la fibrosis intersticial y la de la HVI. Por último, nuestro grupo ha descrito que la PA sistólica y la presión de pulso eran superiores en los pacientes con fibrosis severa que en los pacientes con fibrosis mínima y moderada3.
Factores no hemodinámicos
Dos tipos de hallazgos indican que, además de los factores hemodinámicos, los factores no hemodinámicos también pueden contribuir al desarrollo de la fibrosis miocárdica en la HTA. El primero hace referencia, tal como se ha mencionado anteriormente, a la presencia de fibrosis miocárdica no sólo en el ventrículo izquierdo, sino también en el ventrículo derecho4,12, en el septo interventricular13 y en la aurícula izquierda14, como se ha descrito en estudios post mortem de corazones de pacientes con CH. En segundo lugar, estudios recientes han demostrado que la capacidad del tratamiento antihipertensivo para revertir la fibrosis miocárdica en los pacientes hipertensos es independiente de su eficacia antihipertensiva15,16. Por tanto, la visión actual es que el desarrollo de la fibrosis miocárdica puede ser consecuencia también del predominio de los factores humorales que estimulan sobre los factores humorales que inhiben el metabolismo del colágeno fibrilar (tabla 1)7. Entre aquellos destacan los agonistas del sistema renina-angiotensina-aldosterona (SRAA).

En efecto, en estudios clínicos en los que se inhibía la enzima de conversión de la angiotensina (ECA)15,17 o se bloqueaba el receptor de la angiotensina II (ANG II) de tipo 1 (r-AT1)16,18 se ha puesto de manifiesto la importancia de la ANG II en el desarrollo de la fibrosis miocárdica en los pacientes con CH. De hecho, abundantes estudios experimentales demuestran que la interacción de este péptido con el r-AT1 ejerce múltiples efectos profibróticos en el corazón, incluidas la inducción de hiperplasia de los fibroblastos y la diferenciación de los cardiomiocitos a miofibroblastos, la activación de la síntesis de colágeno fibrilar y la inhibición de la degradación de las fibras de colágeno19. Por otro lado, diversos hallazgos indican que las interacciones entre factores producidos por los cardiomiocitos (p. ej., osteoponina), los macrófagos (p. ej., inhibidor del activador del plasminógeno 1) y los fibroblastos (p. ej., factor de crecimiento transformante beta) mediarían los efectos profibróticos de la ANG II20. Además, la fibrosis podría formar parte de una respuesta reparadora ante la inflamación y el estrés oxidativo inducidos por la ANG II a través de la interacción con el r-AT1 localizado en las células de la microvasculatura cardiaca21.
Otro factor humoral que puede ser importante en el desarrollo de fibrosis miocárdica es la aldosterona. La infusión crónica de aldosterona a ratas uninefrectomizadas y alimentadas con una dieta rica en sodio se asocia con una marcada acumulación de fibras de colágeno en ambos ventrículos cardiacos22. La fibrosis cardiaca en este modelo se previene con espironolactona, un bloqueador del receptor mineralocorticoide23, por lo que el mecanismo fibrótico de la aldosterona comportaría su interacción con dicho receptor presente en los fibroblastos cardiacos y en los miofibroblastos24. Además, la activación del receptor mineralocorticoide puede facilitar las acciones profibróticas de la ANG II a través de la regulación al alza de la expresión de los r-AT125. Es interesante señalar que las acciones profibróticas de la aldosterona parecen ser independientes de la PA, puesto que el bloqueo del receptor mineralocorticoide con eplerenona reduce la fibrosis miocárdica que se produce en ratones con una sobrecarga crónica de presión en ausencia de cambios significativos de la PA sistémica26.
Factores genéticos y ambientales
Algunos hallazgos señalan que los factores genéticos tienen un papel en la modulación de la fibrosis miocárdica hipertensiva. Se ha identificado un marcador microsatélite para el gen de la ECA en la rata, que permite diferenciar los alelos de dicho gen y su asociación con diferentes grados de actividad de la enzima en el plasma27. En las ratas que portan el alelo B se han observado grados más altos de actividad del ECA en el ventrículo izquierdo y un desarrollo de fibrosis ventricular más extenso en respuesta al isoproterenol que en las ratas que portan el alelo L tratadas con el mismo compuesto28. Por otro lado, en un estudio reciente29, nuestro grupo analizó la influencia del polimorfismo A1166C del gen del r-AT1 sobre la capacidad del losartán para inhibir la síntesis del colágeno de tipo I y para revertir la fibrosis miocárdica en pacientes con CH. Los pacientes fueron genotipificados para este polimorfismo y divididos en 2 subgrupos: AA y AC/CC. La síntesis de colágeno era significativamente mayor y disminuía más con el tratamiento con losartán en los pacientes AA que en los pacientes AC/CC. Aunque las bases moleculares de esta asociación no están muy claras, es posible que estén relacionadas con cambios en la actividad cardiaca del SRAA.
Del mismo modo que se sabe que la ingesta exagerada de sodio facilita el desarrollo de la HVI en los animales y los humanos con HTA, independientemente de sus efectos sobre la PA, hallazgos experimentales recientes indican que ello podría ser aplicable también a la fibrosis miocárdica. En efecto, en un estudio reciente se observó que el incremento en la ingesta de sodio se acompañaba del desarrollo de fibrosis miocárdica biventricular en las ratas SHR, pero no en las ratas normotensas Wistar-Kyoto; además, no había asociación entre la elevación de la PA y el aumento de la FVC en las ratas SHR30. Estas observaciones ponen de manifiesto el potencial fibrótico miocárdico de la interacción entre el sustrato genético hipertensivo y los factores exógenos comúnmente ligados a la HTA.
CONSECUENCIAS CLINICAS
Como se muestra en la figura 2, la fibrosis miocárdica puede contribuir a la disfunción ventricular, a la disminución de la reserva coronaria y a las arritmias ventriculares que afectan adversamente al pronóstico cardiovascular de los pacientes con CH31.

Fig. 2. Mecanismos por los que la fibrosis miocárdica contribuye a las complicaciones de la cardiopatía hipertensiva.
Disfunción ventricular
El contenido de colágeno del miocardio le confiere una resistencia al estiramiento (rigidez) durante la diástole32. Asimismo, la elastancia del miocardio durante la contracción depende inversamente del contenido de colágeno32. Diversos estudios clínicos y experimentales han demostrado que la fibrosis incrementa la rigidez y reduce la elastancia del tejido miocárdico33. En concreto, se admite que un incremento del doble o del triple en la FVC influye de manera adversa en la rigidez diastólica (favoreciendo la disfunción diastólica), mientras que un aumento de 4 veces o más de la FVC se asocia con un aumento adicional de la rigidez diastólica y una disminución de la elastancia sistólica (favoreciendo la disfunción sistólica).
Varios hallazgos clínicos apoyan este planteamiento. Recientemente, nuestro grupo ha demostrado que hay una asociación directa entre el contenido de colágeno miocárdico y la rigidez de la cámara ventricular izquierda en pacientes con CH (fig. 3A), y que la regresión de la fibrosis severa inducida por el losartán en estos pacientes se acompaña de la disminución de la rigidez miocárdica18. Sugihara et al34 hallaron que la FVC era el factor que más significativamente se relacionaba con la disfunción diastólica en los pacientes hipertensos, y Brilla et al15 observaron que la disminución de la FVC tras el tratamiento crónico con el inhibidor de la ECA lisinopril se acompañada de la mejoría en la función diastólica ventricular izquierda en pacientes con HTA. Tanto nuestro grupo35 (fig. 3B) como el de McLenachan y Dargie36 han descrito una asociación inversa entre la FVC y la fracción de eyección (FE) en los pacientes con CH. Finalmente, en estudios efectuados en pacientes con insuficiencia cardiaca atribuible a la CH se ha observado que hay una asociación entre la reducción de la fibrosis miocárdica y la mejoría de la función cardiaca de los pacientes37.
Fig. 3. A. Correlación directa de la fracción de volumen de colágeno (FVC) con la rigidez de la cámara ventricular izquierda (KVI), en los pacientes con cardiopatía hipertensiva. B. Correlación inversa de la fracción de volumen de colágeno (FVC) con la fracción de eyección (FE) en los pacientes con cardiopatía hipertensiva. Panel A tomado de Díez et al18. Panel B tomado de Querejeta et al35.
Disminución de la reserva coronaria
Los pacientes con CH pueden presentar síntomas y signos de isquemia miocárdica, aunque angiográficamente las arterias coronarias sean normales38. La reducción de la reserva coronaria es, probablemente, la causa de la isquemia miocárdica en esos casos39.
En la CH se han descrito alteraciones funcionales y estructurales de la microcirculación coronaria que pueden estar relacionadas con la disminución de la reserva coronaria, incluidas la disfunción endotelial, el engrosamiento de la capa media con reducción de la luz y la acumulación de fibras de colágeno en la región periarteriolar40. Se ha publicado que el tratamiento crónico con el inhibidor de la ECA perindopril induce en los pacientes hipertensos un incremento de la reserva coronaria que se asocia con una regresión significativa de la fibrosis periarteriolar y con una ligera, aunque no significativa, reducción de la hipertrofia de la capa media arteriolar18.
La importancia del colágeno periarteriolar en el compromiso de la reserva coronaria queda demostrado por los estudios experimentales de Isoyama et al41, en los que se observó que la normalización de la PA después de desclampar la aorta indujo la regresión de la hipertrofia de la capa media arteriolar, pero la normalización de la reserva coronaria sólo se consiguió tras la inhibición de la acumulación de colágeno en la adventicia con β-aminopropinitrilo. Por tanto, se puede asumir que la fibrosis perivascular es un factor limitante de la distensibilidad de los vasos intramiocárdicos en los pacientes con CH.
Arritmias ventriculares
Estudios epidemiológicos, como el de Framingham42, han puesto de manifiesto una alta incidencia de arritmias ventriculares en los pacientes con CH. Las arritmias se asocian con una mayor mortalidad en estos pacientes, incluida la muerte súbita.
McLenachan y Dargie36 analizaron las posibles correlaciones de las arritmias ventriculares en pacientes con CH y encontraron que los pacientes con arritmias tenían valores superiores de masa ventricular izquierda y FVC que los pacientes sin arritmias. La FE y el número de vasos coronarios con estenosis significativa (> 50%) eran similares en los 2 grupos de pacientes. Por tanto, la alta incidencia de arritmias en pacientes con CH no puede atribuirse excluyentemente a la coexistencia de enfermedad arterial coronaria, o a la disfunción ventricular, y puede estar relacionada con la fibrosis y con los cambios fenotípicos adaptativos de los cardiomiocitos asociados con la HVI.
La fibrosis podría causar arritmias tanto a través de un desacoplamiento anatómico debido a la heterogeneidad miocárdica como a través de un mecanismo de reentrada generado por la propagación en zigzag de la onda transversa43.
ENFOQUE DIAGNOSTICO
Debido a los efectos adversos cardiacos que la fibrosis miocárdica puede tener para los pacientes con CH, su evalución puede ser de ayuda en el diagnóstico del remodelado miocárdico de estos pacientes, así como en el establecimiento de su pronóstico. Además, poder evaluar la fibrosis puede ser útil al valorar los efectos del tratamiento antihipertensivo sobre el remodelado.
Diagnóstico histológico
Dado que la fibrosis miocárdica es una lesión histopatológica, el método diagnóstico más fiable sería la biopsia endomiocárdica. En términos generales, el procedimiento de la biopsia endomiocárdica no presenta complejidad técnica y es clínicamente seguro para el paciente44. Esta afirmación es especialmente relevante si tenemos en cuenta que se ha demostrado que la fibrosis existente en el tabique interventricular es representativa de la que hay en la pared libre del ventrículo izquierdo13, por lo que biopsiar el tabique desde el ventrículo derecho tras acceder a él por vía venosa disminuye aún más el riesgo de complicaciones. No obstante, hay que reconocer que la biopsia endomiocárdica es una metodología invasiva que, por sus exigencias técnicas, presenta limitaciones obvias para su aplicación a gran escala.
Diagnóstico por métodos de imagen
La caracterización ultrasónica del tejido miocárdico permite la identificación y la caracterización del estado físico de éste a través del análisis de las interacciones entre los ultrasonidos y el propio tejido. Esta técnica está basada en el principio de que la interacción de las ondas de ultrasonido con el tejido normal da lugar al reflejo de unas señales ultrasónicas que presentan unas determinadas características que se pueden cuantificar (p. ej., mediante el tissue-integrated backscatter), lo que permite definir la estructura y las cualidades funcionales del miocardio. Estas características difieren de las que se observan cuando los ultrasonidos interactúan con tejido anormal, como el fibroso45. En este contexto, recientemente se ha mostrado una asociación entre las alteraciones en la ecorreflectividad, en forma de disminución de la variación cíclica de la señal de backscatter, y el incremento de la FVC en el corazón de los pacientes con CH46,47. Sin embargo, la reproducibilidad de la información proporcionada por esos métodos es cuestionable. Además, debido a su alto coste, su uso en la práctica clínica habitual estaría restringido a los centros en los que dichos métodos estuvieran disponibles.
La resonancia magnética (RM) es otra técnica prometedora para la caracterización de la composición del miocardio, especialmente la caracterización obtenida con gadolinio, que se ha validado y parece ser útil para la cuantificación de la fibrosis no isquémica. En un estudio realizado en pacientes con miocardiopatía arritmogénica ventricular derecha se ha observado una correlación entre los parámetros medidos con esta técnica y la fibrosis miocárdica, determinada histológicamente48. También se ha demostrado la utilidad de la RM para detectar la fibrosis presente en la enfermedad endomiocárdica49.
Diagnóstico bioquímico
En los últimos años se han desarrollado métodos alternativos a los anteriores basados en la determinación inmunoquímica de los péptidos derivados del metabolismo del colágeno de tipo I y de tipo III presentes en la sangre (tabla 2). De todos los péptidos estudiados, sólo uno, el propéptido carboxiterminal del procolágeno de tipo I (PICP), reúne los requisitos para considerarlo tanto un índice circulante de la síntesis cardiaca de colágeno de tipo I como un biomarcador de la fibrosis miocárdica50.

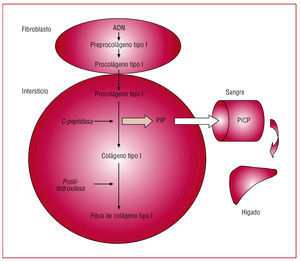
El fundamento de este método es el siguiente (fig. 4): los fibroblastos y los miofibroblastos cardiacos secretan al espacio intersticial la molécula precursora procolágeno de tipo I. Dicho precursor es convertido en la molécula final formadora de fibra, colágeno de tipo I, merced a la acción de proteinasas específicas que hidrolizan los péptidos terminales del precursor. En concreto, una carboxipeptidasa específica hidroliza el PICP que, a través del sistema venoso y el sistema linfático cardíacos, es vertido a la circulación sistémica. Por cada molécula de procolágeno de tipo I que se transforma en una molécula de colágeno de tipo I aparece una molécula de PICP en la sangre, donde se puede detectar mediante un RIA o un ELISA específicos.

Fig. 4. Formación y paso a la sangre del propéptido carboxiterminal del procolágeno tipo I (PICP) liberado durante la conversión del procolágeno de tipo I en colágeno de tipo I. Tomada de López et al50. PICP: propéptido carboxiterminal del procolágeno tipo I.
En estudios piloto realizados por nuestro grupo se ha demostrado que la concentración sérica de PICP se encuentra anormalmente incrementada en pacientes con CH3 y en las ratas SHR51, y que en ambos casos la concentración sérica de dicho péptido se correlaciona directamente con la FVC (fig. 5A). Recientemente hemos demostrado que la concentración de PICP aumenta de manera progresiva con la evolución de la CH, alcanzándose la concentración más elevada en los pacientes con IC y existiendo una correlación directa con el contenido miocárdico de colágeno de tipo I35. Además, en este mismo estudio hemos probado el origen cardiaco del PICP, dado que en los pacientes hipertensos hay un gradiente entre los valores de PICP medidos en sangre coronaria y los determinados en sangre periférica (fig. 5B), así como una correlación directa y altamente significativa entre ambos. Finalmente, en otros estudios hemos descrito que los valores de PICP y la cuantía de la fibrosis miocárdica se modifican paralelamente con el tratamiento antihipertensivo, tanto en ratas SHR7 como en pacientes con CH y función cardiaca normal16,18, y en pacientes con CH e IC37. Aunque preliminares, estos datos indican que el PICP presente en la sangre periférica de los pacientes con CH es esencialmente de origen cardiaco y que se trata de un índice fiable de la cuantía de fibras de colágeno de tipo I presentes en el miocardio, así como de los cambios inducidos por el tratamiento en la cuantía de la fibrosis.

Fig. 5. A. Correlación directa de la fracción de volumen de colágeno (FVC) con la concentración sérica del propéptido carboxiterminal del procolágeno de tipo I (PICP) medido en sangre periférica. B. Correlación directa de la concentración sérica del PICP medido en seno coronario y en sangre periférica. Panel A tomado de Querejeta et al3. Panel B tomado de Querejeta et al35.
Del mismo modo que los péptidos natriuréticos cerebral y atrial se consideran biomarcadores de disfunción sistólica en los pacientes con insuficiencia cardiaca, sería interesante explorar si el PICP puede tener valor como biomarcador de daño estructural miocárdico en esos mismos pacientes. Así se infiere de la observación de que la concentración sérica de PICP es significativamente superior en los pacientes con CH e insuficiencia cardiaca y FE deprimida que en los pacientes con FE preservada52. Sin embargo, dicha diferenciación no sería válida para pacientes con insuficiencia cardiaca de otras etiologías, principalmente cardiopatía isquémica53.
ASPECTOS TERAPEUTICOS
Es posible que haya llegado el momento de proponer que el tratamiento dentes hipertensos no se centre exclusivamente en la normalización de la PA, si no que tenga también como objetivo prevenir o corregir las alteraciones estructurales y funcionales de los órganos diana de la HTA. En relación con la CH, la Sociedad Europea de Hipertensión y la Sociedad Europea de Cardiología establecen en sus guías para el tratamiento de la hipertensión que «los estudios futuros deberían investigar los efectos inducidos por tratamiento sobre el contenido de colágeno y la fibrosis en la pared del ventrículo izquierdo, en lugar de centrarse sólo en la masa»54. Desde esta perspectiva, los fármacos con capacidad para reparar la fibrosis miocárdica serán, fundamentalmente, los que restablezcan el equilibrio entre los factores que estimulan y los factores que inhiben el metabolismo de las moléculas de colágeno de tipo I y de tipo III55.
Hallazgos con fármacos antihipertensivos
Este concepto cardiorreparativo se ha probado clínicamente en varios estudios prospectivos de tamaño limitado en los que se han utilizado biopsias para cuantificar la fibrosis miocárdica. Brilla et al15 demostraron que el tratamiento de pacientes con CH con lisinopril reduce la fibrosis miocárdica independientemente del control de la PA y la regresión de la HVI, mientras que el tratamiento con hidroclorotiacida no tiene tal efecto. La disminución de la fibrosis se asocia con la mejoría de la función diastólica del ventrículo izquierdo. Nuestro grupo ha demostrado que un año de tratamiento con losartán se asocia con la disminución de la concentración sérica de PICP y con la reducción de la FVC en los pacientes con CH18. Sin embargo, los pacientes tratados con amlodipino no muestran cambios significativos en ninguno de los 2 parámetros, a pesar de una eficacia antihipertensiva similar16. En un estudio posterior hemos observado que la capacidad del losartán para inducir la regresión de la fibrosis severa en los pacientes con CH es independiente de su capacidad para reducir la PA o la masa ventricular izquierda, y se asocia con la disminución de la rigidez de la cámara ventricular18. En conjunto, estos datos confirman lo observado en las ratas SHR, en las cuales se ha comprobado que la interferencia farmacológica de las acciones y la producción de ANG II es efectiva en la regresión de la fibrosis cardiaca, con independencia de su efecto antihipertensivo8,56.
Recientemente, hemos descrito que los pacientes con CH e IC tratados de manera crónica con torasemida presentan una disminución mayor de la concentración sérica de PICP y de la FVC, y una mejoría mayor de la clase funcional que los pacientes tratados con furosemida (fig. 6)37. Es interesante señalar que, a diferencia de la furosemida, la torasemida posee la capacidad de inhibir la secreción adrenal de aldosterona57 y su unión al receptor mineralocorticoide58, así como de disminuir la extracción cardiaca de aldosterona en los pacientes con insuficiencia cardiaca59. A partir ello podría hipotetizarse que sus efectos sobre la fibrosis cardiaca se ejercen a través de una cierta inhibición de las acciones profibróticas de la aldosterona.

Fig. 6. Efectos del tratamiento crónico con torasemida o con furosemida sobre la fracción de volumen de colágeno (FVC), la concentración sérica del propéptido carboxiterminal del procolágeno de tipo I (PICP), y la clase funcional (según la clasificación de la New York Heart Association) en pacientes con insuficiencia cardiaca. Los datos se presentan como media ± error estándar de la media. Las columnas oscuras y las claras representan los valores antes y después del tratamiento, respectivamente. Adaptada de López et al37.
Hallazgos con otros compuestos
En diversos trabajos experimentales se han examinado estrategias terapéuticas alternativas para reducir la fibrosis miocárdica. Así, se ha demostrado que el tranilast (ácido N-[3,4-dimetiltoxicinamoil] antranílico)60 y el Ac-SDKP (N-acetil-seril-aspartil-lisil-prolina)61 reducen la inflamación y la fibrosis cardiacas en ratas con hipertensión experimental, a través de mecanismos probablemente relacionados con la inhibición del TGF-β. También se ha observado que los fenofibratos, activadores del PPAR-α, reducen la fibrosis miocárdica en ratas con hipertensión inducida por mineralocorticoides, probablemente a través de la inhibición de la liberación de mediadores inflamatorios asociados con la vía de señalización del NF-κB62. Finalmente, se ha demostrado que el MG132, un inhibidor del proteasoma, suprime la expresión de moléculas de colágeno en fibroblastos aislados y reduce la fibrosis miocárdica en ratas SHR63. Aunque el conjunto de estos hallazgos abre nuevas perspectivas para el tratamiento de la fibrosis miocárdica, hacen falta estudios adicionales para proponer su uso terapéutico en la CH.
Hallazgos con otros procedimientos
Recientemente, se ha descrito que la resincronización cardiaca reduce la FVC en pacientes con IC de etiología diversa64. En otro estudio recién publicado se describe que los efectos beneficiosos de la resincronización sobre la morfología, la función y el rendimiento cardiacos se asocian con la disminución de la concentración sérica de PICP en los pacientes con IC65. Quedan por aclarar los mecanismos precisos por los que la resincronización reduce la síntesis y el depósito de colágeno de tipo I en el miocardio de estos pacientes.
Por otra parte, en estudios experimentales se ha demostrado que la inyección intracardiaca de células madre mesenquimales humanas reduce la fibrosis en animales con infarto de miocardio66, probablemente a través de la liberación de factores que inhiben el metabolismo del colágeno fibrilar67.
CONCLUSIONES
La fibrosis del miocardio forma parte del sustrato histomorfológico de la CH. Los datos disponibles indican que el SRAA interviene de forma determinante en el desarrollo de la fibrosis miocárdica en la CH. La fibrosis puede contribuir a la transición de la HVI a la insuficiencia cardiaca en los pacientes hipertensos, así como al desarrollo de otras complicaciones propias de la CH. Desde esta perspectiva, en el tratamiento clínico de dichos pacientes debe plantearse algo más que el diagnóstico y la normalización de la HTA y la HVI. Una aproximación más completa sería la de plantear medidas dirigidas también a detectar y tratar la fibrosis miocárdica. Algunas evidencias preliminares indican que la medida del PICP sérico puede ser útil para diagnosticar la fibrosis miocárdica en los pacientes con CH. Por otra parte, ya hay evidencias que demuestran que el objetivo de reducir la fibrosis miocárdica es alcanzable en los pacientes con CH, principalmente mediante el uso de fármacos que interfieren con el SRAA. En conjunto, toda esta información sienta las bases para la realización de ensayos clínicos amplios y de larga duración dirigidos a aclarar si el diagnóstico y la regresión de la fibrosis miocárdica contribuyen a mejorar el pronóstico y la evolución de los pacientes con CH.
Correspondencia: Dr. J. Díez Martínez.
Área de Ciencias Cardiovasculares. Edificio CIMA. Facultad de Medicina.
Pío XII, 55. 31008 Pamplona. Navarra. España.
Correo electrónico: jadimar@unav.es