Varón de 57 años con artropatía de columna y síndrome del túnel carpiano bilateral intervenido se presentó por cuadro de anorexia, pérdida de peso y episodios ocasionales de dolor epigástrico, náuseas y vómitos. En el último año había comenzado con disnea de esfuerzo. La exploración física era anodina.
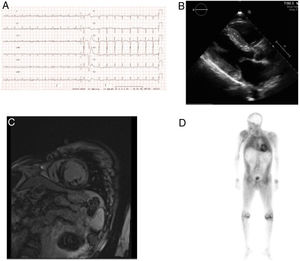
El electrocardiograma mostraba bajos voltajes y el ecocardiograma, hipertrofia del ventrículo izquierdo grave y asimétrica, con predominio septal (19 mm); el ventrículo izquierdo era de tamaño normal, la función sistólica estaba conservada (fracción de eyección del ventrículo izquierdo del 54%) y el patrón de la función diastólica era seudonormal (figura 1A-B).

A: electrocardiograma de bajo voltaje en las extremidades. B: ecocardiograma con hipertrofia ventricular izquierda. C: resonancia magnética cardiaca con realce tardío subendocárdico extenso. D: gammagrafía con 99mTc-ácido 3,3-difosfono-1,2-propanodicarboxílico; mayor captación miocárdica que en hueso.
Con la sospecha de enfermedad por depósito, se realizó un estudio analítico; el hemograma y la bioquímica resultaron normales, sin proteinuria. El patrón electroforético en suero era normal y las cadenas kappa y lamda libres, negativas. La resonancia magnética cardiaca con gadolinio mostró áreas extensas de realce subendocárdico en ambos ventrículos y la aurícula izquierda, compatible con amiloidosis (figura 1C). En el estudio gammagráfico con 99mTc-ácido 3,3-difosfono-1,2-propanodicarboxílico, se observó depósito del radiotrazador en el ventrículo izquierdo, con intensidad mayor que la del hueso (figura 1D). No se detectó amiloide en las biopsias de grasa subcutánea y rectal. Se realizó valoración neurológica y estudio de neuroconducción sensitivo-motora, que no mostró alteraciones, excepto afección del nervio mediano bilateral.
A los 5 meses, el paciente acudió a urgencias en edema agudo de pulmón. En el ecocardiograma el ventrículo izquierdo no dilatado presentaba disfunción moderada (fracción de eyección del ventrículo izquierdo del 38%) y función diastólica con patrón restrictivo. La fracción N-terminal del propéptido natriurético cerebral fue 14,879 pg/ml.
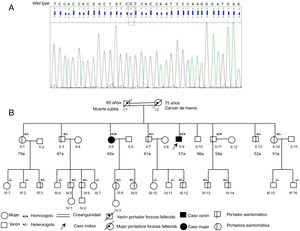
El estudio genético, para determinar si se trataba de amiloidosis por depósito de transtiretina tipo salvaje (wild type) o la forma hereditaria, confirmó mutación en homocigosis Val142Ile (clásicamente Val122Ile) del gen de la transtiretina (figura 2A). En la evaluación familiar (figura 2B) no se conocen ancestros de raza negra; los padres, fallecidos, eran primos. La madre no tenía antecedentes cardiológicos conocidos y el padre tuvo una enfermedad cardiaca sin precisar desde los 40 años y falleció súbitamente a los 65 años. Estudiados 7 de 8 hermanos, se hallaron 2 homocigotos, todos sin síntomas; el electrocardiograma y el ecocardiograma fueron normales en todos, excepto en una de las hermanas con la mutación en homocigosis y electrocardiograma normal, síndrome del túnel carpiano bilateral, ecocardiograma con hipertrofia moderada (septo de 14 mm) y gammagrafía con captación en ventrículo izquierdo similar a la del hueso. Entre los sobrinos se encontraron 8 portadores asintomáticos, con electrocardiograma y ecocardiograma sin alteraciones.

Con el diagnóstico de amiloidosis cardiaca hereditaria por mutación en el gen de la transtiretina, se derivó al paciente a un centro de referencia para trasplante hepático y cardiaco, que se realizaron con éxito.
Las mutaciones en la transtiretina constituyen la causa más frecuente de amiloidosis, y originan neuropatía y con frecuencia afección cardiaca. Se conocen múltiples mutaciones que generan diferentes fenotipos1.
La mutación encontrada (Val122Ile) produce amiloidosis cardiaca en mayores de 60 años con fenotipo similar a transtiretina wild type, asociada ocasionalmente con síndrome del túnel carpiano. Entre un 3 y un 4% de los individuos de raza negra en Estados Unidos son portadores heterocigotos de esta mutación2, que es rara en población blanca. Pese a que se ha considerado una mutación bastante indolente, con cardiomiopatía a edad tardía, varios estudios la relacionan con mayor morbimortalidad respecto a la forma salvaje3. Si se considera la presentación en homocigosis, este riesgo aumenta y aparece insuficiencia cardiaca más precoz y grave4.
Nuestro paciente manifestó un fenotipo grave, con rápida progresión a insuficiencia cardiaca, clase funcional II/IV de la New York Heart Association, disfunción ventricular izquierda e importante elevación de la fracción N-terminal del propéptido natriurético cerebral.
El estudio genético no solo fue de utilidad en el diagnóstico de amiloidosis hereditaria, sino que ayudó a comprender la rápida evolución, al encontrarse en homocigosis.
Se han desarrollado nuevos tratamientos con el objetivo de detener o retrasar el depósito amiloideo por transtiretina que actúan a diferentes niveles5. Algunos ya han mostrado su efectividad en ensayos clínicos aleatorizados y han sido aprobados por las agencias reguladoras. Algunos de estos fármacos ya aprobados actúan inhibiendo la expresión hepática de transtiretina mediante ácido ribonucleico de interferencia (patisirán) o mediante oligonucleótidos antisentido (inotersén). Otros fármacos actúan estabilizando la molécula de transtiretina e impidiendo su disociación y depósito. A este grupo pertenece tafamidis, del que se ha demostrado reducción en la mortalidad y disminución de ingresos de causa cardiovascular6. Existen otros estabilizadores en desarrollo. Por último, se espera que sea posible eliminar depósitos de amiloide mediante anticuerpos dirigidos contra la transtiretina o mediante moléculas como la doxiciclina. Actualmente están en marcha varios ensayos que evalúan estos compuestos5.
El cribado genético permitió identificar a 15 portadores que requieren seguimiento estrecho y se podrán beneficiar de los nuevos tratamientos precozmente, para frenar el desarrollo de la enfermedad y mejorar el pronóstico.
En nuestro caso un diagnóstico temprano habría permitido iniciar el tratamiento farmacológico eficaz y evitar la fase terminal que condujo al trasplante. Afortunadamente los portadores identificados, en especial la hermana con la enfermedad en fase precoz, se beneficiarán de los tratamientos disponibles.