Palabras clave
INTRODUCCION
El síndrome de QT largo (SQTL) se caracteriza por una grave alteración en la repolarización ventricular traducida en el electrocardiograma (ECG) por un alargamiento en el intervalo QT que predispone a arritmias ventriculares malignas --torsade de pointes-- y muerte súbita. Fue descrito clínica y electrocardiográficamente en 19571 por Anton Jervell y Fred Lange Nielsen, quienes publicaron sus estudios en una familia de progenitores no consanguíneos con 6 hijos, 4 de los cuales tenían sordera congénita y episodios sincopales, y 3 de ellos tuvieron muerte súbita. El ECG de los casos mostraba un intervalo QT inusualmente largo. Ambos progenitores estaban asintomáticos, tenían un ECG normal y no presentaban problemas de audición. En 1964, Romano y Ward publicaron de forma independiente un síndrome cardiaco familiar caracterizado por síncope recurrente, antecedente familiar de muerte súbita y prolongación del intervalo QT sin sordera neuronal2. Los estudios genéticos posteriores mostraron que el síndrome descrito por Jervell y Lange Nielsen, que se acompaña de sordera neuronal congénita, corresponde a mutaciones homocigotas, con un fenotipo muy grave y alto riesgo de muerte súbita. Por otro lado, el síndrome conocido como Romano-Ward corresponde a mutaciones generalmente heterocigotas, los pacientes no presentan trastornos en la audición y la gravedad de la enfermedad es muy variable. Después de casi medio siglo, en 19953,4 se describieron los principales genes asociados con la enfermedad y el SQTL se reconoció por primera vez como una canalopatía cardíaca; fue la primera en ser descrita como tal y es, quizá, la canalopatía arritmogénica mejor estudiada hasta el momento. El cuadro clínico es muy variable: el paciente puede cursar asintomático, presentar síncope recurrente, crisis convulsivas o muerte súbita como primera manifestación de la enfermedad. Inicialmente se catalogó como un trastorno raro y, en efecto, la presentación grave de la enfermedad es esporádica, pero la incidencia de las mutaciones se estima en 1/3.000-5.000 casos5, cerca del 32% de los portadores asintomáticos puede tener un QT corregido por frecuencia (QTc) en los límites normales, transmiten la enfermedad a un 50% de su descendencia, son más susceptibles a desarrollar arritmias malignas comparados con el resto de la población y hasta un 20% puede volverse sintomático6.
El SQTL presenta gran heterogeneidad genética y se han identificado ya más de 500 mutaciones distribuidas hasta ahora en 10 genes: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 y SCN4B. A pesar de los avances en la materia, un 25-30% de los pacientes permanece sin diagnóstico genético7,8. Es una enfermedad de presentación principalmente monogénica6; las variedades poligénicas o compuestas suelen dar un fenotipo más grave. La penetrancia --casos que tienen la mutación y el fenotipo-- oscila entre el 25 y 90%9 y con menos frecuencia puede haber variaciones en la expresividad de la enfermedad --diversos fenotipos que puede dar una misma mutación.
Los estudios genéticos moleculares desarrollados en los últimos 11 años han permitido realizar una importante correlación genotipo-fenotipo y orientar así el tratamiento; también se han hecho interesantes observaciones en cuanto a susceptibilidad individual a desarrollar arritmias al estudiar los efectos de polimorfismos no sinónimos frecuentes en la población, lo que ha motivado gran interés, sobre todo en el área de la farmacogenómica.
CLASIFICACION DEL SINDROME DE QT LARGO
Conceptos generales
La clasificación utilizada en el pasado se basa en la presentación homocigota o heterocigota de la enfermedad, que dan lugar a los síndromes de Jervell-Lange-Nielsen (con sordera) y Romano Ward (sin sordera), respectivamente. La clasificación actual enfatiza los hallazgos genéticos, como se ilustra en la tabla 1. En 1995-1996 se describieron los 3 principales genes asociados con la enfermedad. Codifican para unidades formadoras del poro de los canales de potasio IKs e IKr y de sodio Nav 1,5; explican cerca del 65% de los casos. Si bien en los años subsecuentes se han añadido 7 genes más a la lista, éstos explican tan sólo cerca del 5% de los casos.

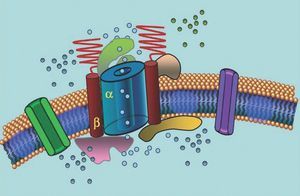
Los canales iónicos son proteínas transmembranales encargadas de transportar iones a través de la membrana celular; los canales implicados en el SQTL son selectivos o especializados en el transporte de un solo ion y dependientes de voltaje, es decir, su activación ocurre a determinado voltaje intracelular (varía según el subtipo de canal). Los fenómenos eléctricos y contráctiles que suceden en el cardiomiocito son controlados por estas estructuras. Los canales iónicos forman complejos macromoleculares; hay una unidad principal formadora del poro del canal y proteínas auxiliares que lo regulan (fig. 1). La afección en la función de un canal en el SQTL se puede dar en estos 2 sitios: en la proteína principal o en las proteínas reguladoras (tabla 1). La afección en la unidad formadora del poro, conocida como alfa, genera los 3 subtipos más comunes de SQTL: SQTL1 (afección en el canal de potasio IKs), SQTL2 (afección en el canal de potasio IKr) y SQTL3 (afección en el canal de sodio). Al ser los más frecuentes, han sido mejor caracterizados clínicamente y genéticamente. En la figura 2 se describe la correlación fenotipo-genotipo de estas 3 principales formas. El síndrome llamado Jervell-Lange-Nielsen corresponde en la actualidad a las variedades de SQTL 1 y 5. De manera característica, los pacientes cursan con sordera congénita y tienen mutaciones homocigotas o heterocigotas compuestas que afectan a la corriente IKs. El síndrome de Romano Ward abarca desde la variedad SQTL 1 hasta la 10 y no cursa con sordera.

Fig. 1. Representación esquemática del complejo macromolecular. Los canales iónicos son proteínas transmembranales (α) reguladas por diversas proteínas, una de ellas es la llamada subunidad β.
Síndrome de QT largo tipo 1 (SQTL1)
Los pacientes con SQTL1 suelen presentar episodios de arritmia ventricular al realizar ejercicio o al estimular el simpático (68%)10; la natación se ha descrito como un deporte disparador de arritmias en el SQTL111. La penetrancia en este subtipo es cercana al 62%. Con frecuencia estos pacientes presentan una onda T de base ancha, con una duración muy prolongada12,13 (fig. 2).

Fig. 2. Correlación genotipo-fenotipo en los síndromes de QT largo más frecuentes. *Se refiere a los casos que tienen la mutación y manifiestan el fenotipo.
Es el subtipo más frecuente y explica 30-35% de los casos. El gen afectado es el KvLQT1 (o KCNQ1), localizado en el cromosoma 11 (11p15.5), codifica la subunidad α, del canal de potasio IKs. El potencial de acción se prolonga por una disminución de la corriente saliente de K+ durante fase 3 del potencial de acción.
Síndrome de QT largo tipo 2 (SQTL2)
Los pacientes con SQTL2 suelen presentan arritmias ventriculares en respuesta al estrés emocional (49%) o estímulos auditivos súbitos --por ejemplo, al reloj despertador-- y con menos frecuencia durante el sueño (22%) o el ejercicio (29%)10. Este subtipo es particularmente susceptible a presentar arritmias en el período posparto14. La penetrancia estimada es del 79% y significa que hasta un 20% de los casos pueden tener un ECG no diagnóstico. En el SQTL2, la onda T suele ser de baja amplitud, bífida, con muescas12,13 (fig. 2). El gen afectado es el KCNH2 o HERG, localizado en el cromosoma 7 (7q35-36), el cual codifica la subunidad α del canal de potasio IKr; explica 25-30% de los casos. La disfunción de este canal disminuye la corriente saliente de K+ durante la fase 3 del potencial de acción, prolongando su duración.
Síndrome de QT largo tipo 3 (SQTL3)
Los casos con SQTL3 tienen un riesgo mayor de presentar arritmias malignas durante el reposo (sueño) o bradicardia15. La penetrancia de las mutaciones en el gen SCN5A es cercana al 90%. El ECG en el SQTL3 suele mostrar onda T acuminada, de aparición tardía, que deja observar con claridad el alargamiento del segmento ST12,13 (fig. 2). Estos pacientes suelen ser menos sintomáticos que los casos con SQTL1 o SQTL2, pero los eventos son característicamente más letales.
El gen afectado en el SQTL3 es el SCN5A, que codifica para la subunidad α del canal de sodio Nav1.5 (fig. 1), localizado en el cromosoma 3 (3p21-24); es causante de la enfermedad en el 5-10% de los casos. La inactivación defectuosa del canal permite la entrada sostenida de Na+ durante la fase 2 del potencial de acción y prolonga su duración.
Síndrome de QT largo tipo 4 (SQTL4)
Es una variedad rara y explica cerca del 1% de los casos. Condiciona un SQTL muy atípico con un gran espectro de arritmias que incluyen taquicardia ventricular polimórfica catecolaminérgica, fibrilación auricular, trastornos en la conducción intraventricular, disfunción del nódulo sinusal y bradicardia16-18; con frecuencia, los casos tienen incluso el QTc en los límites normales. El gen afectado es el ANKB, localizado en el cromosoma 4 (4q25-27), el cual codifica la síntesis de anquirina-β, una proteína estructural que vincula proteínas de la membrana del cardiomiocito con proteínas del citoesqueleto. Estas proteínas son: la bomba Na/K ATP-asa, el intercambiador Na/Ca y el receptor a inositol trifosfato (InsP3R). Las mutaciones que causan pérdida de la función de anquirina-β resultan en un incremento de la concentración de calcio intracelular, así como en una alteración en la expresión de N/K ATP-asa y en el intercambiador Na/Ca. La elevación de las concentraciones de calcio da lugar a posdespolarizaciones tempranas y tardías. De esta manera, las arritmias ventriculares observadas en las mutaciones del gen de anquirina-β se deben a despolarizaciones espontáneas generalmente en respuesta a la estimulación catecolaminérgica.
Síndrome de QT largo tipo 5 (SQTL5)
Está condicionado por cambios de secuencia del gen KCNE1 localizado en el cromosoma 21 (21q22.1-p22)19. Codifica la síntesis de la subunidad β del canal IKs, conocida también como subunidad minK que regula al canal IKs. Explica menos del 1% de los casos.
Síndrome de QT largo tipo 6 (SQTL6)
El gen afectado es el KCNE2 localizado en el cromosoma 21 (21q22.1)20. Codifica la subunidad β del canal de potasio, conocida también como subunidad MiRP1, que regula al canal IKr . Explica menos del 1% de los casos.
Síndrome de QT largo tipo 7 o Andersen-Tawil (SQTL7)
Los hallazgos dismórficos y las alteraciones electrocardiográficas de este síndrome fueron descritos por primera vez en 1971 por el Dr. Andersen21 y recapitulados en 1994 por el Dr. Tawil22, pero la descripción genético-molecular se publicó apenas en el año 200123. El ahora conocido síndrome de Andersen-Tawil (SAT) es una alteración autosómica dominante que se caracteriza por parálisis periódica, desarrollo esquelético anormal, arritmias ventriculares del tipo de la extrasistolia ventricular frecuente con susceptibilidad particular a desarrollar fibrilación ventricular, sobre todo en el sexo femenino. Los trastornos del ritmo descritos en el SAT son: extrasístoles ventriculares (41%), taquicardia ventricular polimórfica no sostenida (23%), taquicardia ventricular bidireccional (68%) y torsades de pointes (3%)24. Algunos de los rasgos dismórficos observados incluyen: estatura corta, escoliosis, clinodactilia, hipertelorismo, implantación baja de orejas, micrognatia y frente amplia. La expresividad de la enfermedad es variable, lo que complica el diagnóstico oportuno23,25. El 70% de los casos se explica por mutaciones en el gen KCNJ2 localizado en el cromosoma 17 (17q23) que codifica la síntesis del canal rectificador de potasio Kir 2,1; este canal participa en la fase 4 del potencial de acción. Varios autores cuestionan la inclusión de este gen dentro del grupo causal de SQTL, pues el intervalo QTc en este síndrome se encuentra ligeramente prolongado o incluso normal, pero la onda U suele ser prominente, lo que ha dado lugar a la sobrevaloración del intervalo QT. El lector encontrará que algunos autores proponen que las mutaciones en KCNJ2 generan el SAT1 y no el SQTL724.
Síndrome de QT largo tipo 8 (SQTL8)
Resulta de mutaciones en el gen CACNA1 que codifica el canal de calcio tipo L Cav1.2, localizado en el cromosoma 12 (12p13.3). Ocasiona el síndrome de Timothy26, caracterizado por malformaciones cardiacas, deficiencia inmunológica, hipoglucemia intermitente, trastornos cognitivos incluido el autismo, fusiones interdigitales y QT largo que predispone a arritmias cardiacas y muerte súbita27. Explica menos del 0,5% de los casos.
Síndrome de QT largo tipo 9 (SQTL9)
Esta variedad resulta de mutaciones en el gen CAV3, localizado en el cromosoma 3 (3p25), que codifica la síntesis de caveolina 3. La caveola es una invaginación de la membrana plasmática implicada en la endocitosis, la homeostasis de lípidos y la transducción de señales. Un importante componente de esta estructura es la caveolina, de la cual se conocen 3 subtipos; el subtipo 3 es específico de músculo esquelético y cardiaco. Algunos canales iónicos se colocalizan en la caveola, incluida la isoforma cardiaca de canal de sodio Nav1.5; recientemente se describieron diversas mutaciones en esta proteína que alteran las propiedades biofísicas del canal de sodio Nav1,5 in vitro, generando un fenotipo similar al observado en el SQTL328. Se estima que explica < 1% de los casos.
Síndrome de QT largo tipo 10 (SQTL10)
Esta variedad fue notificada en un caso muy grave con un QTc > 600 ms, bradicardia fetal y bloqueo auriculoventricular (AV) 2 1. Resulta de mutaciones en el gen SCN4B, localizado en el cromosoma 11 (11q23) que codifica para la subunidad β4 de canal de sodio. Se han descrito 4 distintos subtipos de subunidades β que interaccionan y regulan las diversas isoformas de canal de sodio, pero sólo el subtipo 4 se ha asociado hasta ahora con arritmogénesis29. La incidencia de mutaciones en este gen no ha sido explorada, pero se estima < 1%.
Mutaciones de la variedad Jervell-Lange-Nielsen
Esta grave forma de SQTL está causada por mutaciones homocigotas30 o heterocigotas compuestas en los genes KCNQ1 y/o KCNE1 que codifican la corriente IKs, es decir, se trata de una variedad muy grave de las formas SQTL1 o SQTL5. Se asocia de manera característica con sordera congénita. Los pacientes suelen tener un QTc > 500 ms, síncope recurrente y alto riesgo de muerte súbita. Los progenitores de los pacientes con esta variedad son generalmente heterocigotos y manifiestan una enfermedad menos grave, o incluso pueden ser asintomáticos31.
DIAGNOSTICO DEL SINDROME DE QT LARGO
Índice de puntuación de Schwartz
En 1985, Schwartz et al32 publicaron los criterios diagnósticos para el SQTL, modificados en 1993, que representan una importante guía en la evaluación inicial de los casos potenciales. Utilizan una puntuación del 1 al 9 según la historia familiar y los hallazgos clínicos y electrocardiográficos. Si el índice de puntuación es ≤ 1, la probabilidad de presentar la enfermedad es baja, si es 2-3 la probabilidad es intermedia, y si ≥ 4, es alta (tabla 2).

Diagnóstico prenatal de síndrome de QT largo
La bradicardia fetal puede ser una de las primeras manifestaciones clínicas del SQTL. En series retrospectivas se ha documentado que hasta un 70% de los pacientes diagnosticados en la infancia tiene este antecedente, que suele ir acompañado de hidrops fetalis33. La evaluación de la repolarización cardiaca fetal entre las semanas 14 y 39 es un método útil para el diagnóstico oportuno del SQTL34.
Mosaicismos gonadales para SQTL se han asociado con pérdidas fetales recurrentes durante el tercer trimestre del embarazo35. Si la sospecha de la enfermedad es muy alta, la amniocentesis a partir de las 16 semanas de gestación puede ser de utilidad para el diagnóstico, que resulta sencillo cuando alguno de los progenitores es conocido como portador de una mutación determinada36.
ESTUDIO DEL PACIENTE CON SINDROME DE QT LARGO
Historia clínica
Los antecedentes familiares y/o personales de muerte súbita son de crucial importancia, tanto para el diagnóstico del SQTL como para la estratificación de riesgo. Asimismo, los factores precipitantes y el contexto del síncope pueden señalarnos el subtipo de SQTL. En la valoración inicial de un caso sospechoso se debe descartar el uso de fármacos que pudieran prolongar el intervalo QT.
Intervalo QT: ¿cuánto es lo normal?
El intervalo QT se debe medir de manera preferente en las derivaciones II o V537, donde se ha documentado que tiene mayor poder predictivo38. Traduce la duración de la repolarización ventricular y se mide desde el inicio de la onda Q hasta el final de la onda T. Convencionalmente, se utiliza la fórmula de Bazett39 para corregir la duración del intervalo de acuerdo con la frecuencia cardiaca (QTc = QT/√RR, expresado en segundos). Si bien la medición de este intervalo parece sencilla, en un estudio multicéntrico realizado por Viskin et al40, menos del 40% de los médicos no cardiólogos, menos del 50% de los cardiólogos y más del 80% de los arritmiólogos supieron medirlo correctamente. Es aconsejable que el médico realice una medición manual y no confiar en las mediciones automatizadas que, si bien son útiles para otros intervalos, suelen ser imprecisas en el cálculo del intervalo QT. Es un intervalo dinámico y los límites normales dependen de varios factores. Si bien se ha considerado anormal un intervalo QTc ≥ 440 ms en los varones y ≥ 460 ms en las mujeres, en este rango podemos encontrar tanto a portadores de mutaciones como a sujetos sanos (fig. 3). Vincent et al41 demostraron que, en familias con SQTL1, ningún caso con genotipo positivo tuvo un QTc < 410 ms y ninguno con genotipo negativo tuvo un QTc > 470 ms. Monnig et al38 mostraron recientemente que un QTc > 440 ms es eficaz para detectar a pacientes con mutaciones asociadas SQTL, un QTc > 470 ms es útil para detectar a pacientes en riesgo de desarrollar síntomas, y un QTc > 500 ms se encontró en pacientes sintomáticos en tratamiento.

Fig. 3. Modelo de distribución de intervalo QT corregido por frecuencia (QTc) en pacientes con mutaciones en KVLQT1, HERG, or SCN5A y sus familiares no afectados. La curva de la izquierda describe la distribución de los no afectados y la de la derecha la de los afectados.
Otras alteraciones electrocardiográficas asociadas al síndrome de QT largo
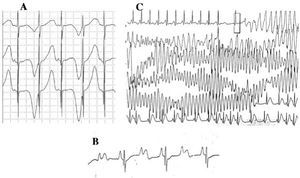
Los pacientes con SQTL pueden presentar múltiples alteraciones en la onda T: alternancia en la polaridad, apariencia bifásica, variaciones en la amplitud, muescas, entre otras42. La alternancia de la onda T (fig. 4A) se define como la variación latido a latido de la amplitud, la morfología y la polaridad de la onda T en ritmo sinusal, sin variaciones en el complejo QRS. Constituye un indicador de inestabilidad eléctrica43, refleja dispersión regional en la repolarización ventricular y en ocasiones precede a la fibrilación ventricular44.

Fig. 4. Alteraciones electrocardiográficas en el síndrome del QT largo. A: alternancia eléctrica de la onda T. B: bloqueo auriculoventricular 2:1. C: torsade de pointes autolimitada.
Los pacientes con SQTL pueden cursar con signos de disfunción del nódulo sinusal, bradicardia y/o pausas45. Los subtipos SQTL1 y SQTL3, particularmente este último, presentan con frecuencia bradicardia sinusal46, mientras el SQTL4 se ha asociado con disfunción del nódulo sinusal18.
Desde las décadas de 1970-1980 se observó la coexistencia de trastornos en la conducción AV con el SQTL47 (fig. 4B). El bloqueo AV 2:1 es una manifestación infrecuente pero de mal pronóstico, que puede presentarse desde la etapa fetal en forma de bradicardia persistente. La incidencia ha sido comunicada en el 4-5%48 y se asocia con una alta mortalidad a pesar del tratamiento con bloqueadores beta y/o marcapasos49,50. Este fenómeno puede explicarse por la exagerada duración del potencial de acción. Al alargarse el período refractario ventricular, el siguiente impulso procedente de la actividad sinusal es bloqueado por encontrar a los ventrículos aún en período refractario. Esta alteración parece ser particular del SQTL, pues el período refractario ventricular es mayor que en el sistema de conducción AV51. El complejo QRS es usualmente angosto y el bloqueo se ha localizado en la zona infrahisiana46,51,52, pero el sitio de bloqueo podría depender del genotipo. Hasta el momento, 4 genes han sido relacionados a bloqueo 2:1 en el contexto de SQTL: HERG(SQTL2)53,54, SCN5A(SQTL3)52, CACNA1 (SQTL8)26 y SCN4B(SQTL10)55.
La arritmia ventricular característica del SQTL es la conocida torsade de pointes (fig. 4C). Se presenta cuando el intervalo QT se prolonga, independientemente de la etiología. Es una taquicardia ventricular polimórfica por reentrada, caracterizada electrocardiográficamente por un giro continuo del eje del QRS sobre una línea imaginaria. Suele estar precedida de una pausa seguida de una extrasístole --intervalo RR «corto-largo-corto»--, como se muestra en la figura56-58. Puede culminar en fibrilación ventricular y muerte súbita. Si esto no sucede, el paciente puede experimentar sólo un síncope o incluso, si el episodio es breve, puede pasar desapercibido.
Holter
El estudio Holter permite una valoración amplia y dinámica del intervalo QT; en ocasiones pueden registrarse episodios espontáneos de arritmia ventricular asintomática, así como posibles episodios de disfunción del nódulo sinusal o bloqueo AV.
Prueba de esfuerzo
Los pacientes con SQTL no suelen alcanzar la frecuencia máxima esperada calculada para la edad. Asimismo, el intervalo QT al esfuerzo puede tener un comportamiento paradójico, alargándose en lugar de acortarse59,60. El comportamiento electrocardiográfico durante la prueba de esfuerzo será diferente según el subtipo SQTL. Los pacientes con SQTL1, además de no llegar a la frecuencia cardiaca máxima calculada para la edad, con frecuencia alargan el intervalo QT, mientras que los que tienen un SQTL2 suelen alcanzar la frecuencia cardiaca esperada y prolongar sólo discretamente el intervalo QT, o incluso no prolongarlo61,62. Los pacientes con SQTL3 tienen, en general, una respuesta fisiológica al ejercicio, esto es, un acortamiento normal del intervalo QT63. Este estudio también puede ser útil para valorar la respuesta al tratamiento y estratificar el riesgo en los casos asintomáticos o que presentan dudas acerca de los factores precipitantes de las arritmias.
Tamizaje genético
El estudio genético en el SQTL ha sido limitado el los últimos años a laboratorios de investigación; sin embargo, la información derivada de éste es de gran utilidad en el tratamiento de los enfermos, en particular en los casos de alto riesgo. La principal aplicación es, quizá, el consejo genético, pero también tiene importantes implicaciones en el tratamiento, que puede orientarse según el canal afectado. La localización precisa de una mutación dada puede otorgar información adicional en cuanto a la evaluación del riesgo. Las mutaciones en la región transmembranal de KCNQ1 (IKs) tienen mayor probabilidad de presentar eventos arrítmicos que las mutaciones en la región C-terminal64, así como las mutaciones en la región del poro de KCNH2 o HERG65 comparadas con mutaciones en la región N o C-terminal66.
El escrutinio inicial quizá se pueda limitar a los genes KCNQ1, HERG y SCN5A, con posibilidad de encontrar mutaciones en un 65% de los casos; en caso de obtener resultados negativos se puede ampliar el tamizaje a los genes KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 Y SCN4B, lo que permitirá incrementar la posibilidad de resultados positivos en un 5-10%.
Tamizaje genético post mortem
Interesante ha sido el hallazgo de mutaciones en genes que condicionan SQTL en casos de niños con muerte súbita infantil y en casos de muerte súbita inexplicable en el adulto joven.
El estudio genético post mortem en pacientes con muerte súbita y autopsia negativa ha mostrado mutaciones que condicionan SQTL en porcentajes variables67-69 cercanos al 10% en la muerte súbita infantil y al 35% en la muerte súbita del adulto joven70-72. Sobre la base de estos resultados, se ha propuesto realizar un ECG sistemático en todos los neonatos73,74.
El estudio genético post mortem, también conocido en la literatura científica como «autopsia molecular», además de tener repercusiones legales, tiene importantes implicaciones en los familiares de los casos que pudieran estar afectados sin saberlo.
Polimorfismos reguladores
Se han descrito diversos polimorfismos frecuentes en la población, distribuidos en prácticamente todos los genes asociados al SQTL. Si bien estos cambios no son aparentemente patogénicos, algunos pueden tener los siguientes efectos75-78:
1. Generar susceptibilidad individual para desarrollar arritmias.
2. Favorecer el efecto patogénico de otro cambio no sinónimo.
3. Disminuir el efecto patogénico de otro cambio no sinónimo.
Como el polimorfismo K897T en KCNH2 (HERG), con una frecuencia en la población de hasta un 15%, que no sólo se ha asociado con susceptibilidad a determinados fármacos79, también favorece el efecto patogénico de mutaciones en el mismo gen78. Otro ejemplo es el polimorfismo S1103Y en el gen SCN5A encontrado principalmente en la población de raza negra, con una incidencia cercana al 13% y asociado con un incremento en el riesgo de muerte súbita infantil80.
Interesante también ha sido la descripción de dos sitios de procesamiento alternativo en el producto del gen SCN5A que codifica la isoforma de canal de sodio cardiaco Nav1,5 en el ser humano, que genera 2 tipos de canales de sodio: uno con 2.016 aminoácidos que contiene una glutamina en la posición 1077 (Q1077), y otro con 2.015 aminoácidos que carece de esta glutamina (Q1077del). Transcritos de estos 2 procesamientos alternativos están presentes en un mismo corazón humano a razón de 2:1 y diversos polimorfismos frecuentes tendrán un efecto distinto en la función del canal dependiendo del contexto Q1077 o Q1077del. Esto se demostró inicialmente con el polimorfismo H558R de SCN5A, presente hasta en un 30% de la población; al expresar H558R en el contexto de Q1077 se observó una profunda disminución de la corriente iónica81. Un efecto similar se documentó con el polimorfismo S524Y82. Estos hallazgos han dado elementos para explicar la variabilidad de la gravedad de la enfermedad, así como los distintos fenotipos de una misma mutación observados en algunas familias77.
Prueba farmacológica con adrenalina
La prueba farmacológica con adrenalina en dosis bajas es una alternativa útil y segura para desenmascarar los casos sospechosos de SQTL con un QTc limítrofe. Es particularmente eficaz para detectar formas asintomáticas de SQTL1, con una sensibilidad del 92,5% y una especificidad del 86%; un valor predictivo positivo del 76% y un valor predictivo negativo del 96%. Puede ser útil también en el diagnóstico del SQTL2, con menor sensibilidad y especificidad. No es útil para el SQTL3 u otras formas de SQTL. En condiciones normales, la estimulación simpática induce la fosforilación del canal de potasio IKs, optimizando su función y dando lugar a un acortamiento del potencial de acción. En pacientes con SQTL, en particular el tipo 1, se observa una respuesta paradójica a la administración de dosis bajas de adrenalina (0,025-0,2 µg/kg/min) que alargan el intervalo QT más de 30 ms83-86.
PROLONGACION DEL INTERVALO QT Y TORSADE DE POINTES INDUCIDA POR FARMACOS
Gran variedad de fármacos utilizados en diversas especialidades médicas pueden ocasionar el alargamiento del intervalo QT de forma iatrogénica. Incluso, algunos medicamentos han sido retirados del mercado por este indeseable efecto (p. ej., el astemizol y la cisaprida, entre otros; para mayor detalles consultar el portal de internet www.qtdrugs.org)87,88.
Las arritmias ventriculares secundarias a fármacos no antiarrítmicos se presentan en menos de un caso por cada 10.000-100.000 expuestos. Considerando que los estudios clínicos incluyen entre 2.000 y 3.000 sujetos, es fácil que este indeseable y fatal efecto secundario escape a la detección como un efecto adverso en la fase clínica del desarrollo de fármacos89. Este punto ha generado enorme interés en lo que se refiere a aspectos de seguridad en el estudio y la generación de nuevos fármacos.
Los factores relacionados con la susceptibilidad individual son: sexo femenino, hipocalcemia, hipomagnesemia, bradicardia, insuficiencia cardiaca congestiva, poscardioversión, fibrilación auricular, hipertrofia ventricular izquierda, SQTL no detectado, polimorfismos predisponentes y altas concentraciones séricas de los fármacos predisponentes90.
El canal que por excelencia interacciona con fármacos es el IKr, codificado por el gen KCNH2 (HERG). Esto se debe a la estructura molecular de este canal, mientras que otros canales de potasio tienen 2 residuos prolina que se inclinan en forma angulada hacia el poro del canal disminuyendo su lumen. IKr carece de ellos, lo que genera un vestíbulo del poro más amplio y facilita su exposición a grandes moléculas. Por el contrario, tiene dos residuos aromáticos (tirosina y fenilalanina) que facilitan enlaces con moléculas aromáticas presentes en diversos fármacos capaces de bloquear el canal91.
Como mencionamos anteriormente, el SQTL tiene penetrancia incompleta y hay portadores asintomáticos de mutaciones que pueden manifestar arritmias malignas al recibir alguno de estos fármacos. Por otro lado, polimorfismos considerados frecuentes en la población confieren susceptibilidad individual a desarrollar torsades de pointes con el uso de fármacos, como sucede con el polimorfismo R1047L, el segundo observado con más frecuencia en KCNH2 y que se ha asociado con torsades de pointes con el uso del fármaco dofetilida92. Se han descrito por lo menos 20 polimorfismos en sujetos sanos en el gen KCNH2 y su efecto en la susceptibilidad individual a desarrollar arritmias malignas relacionadas con fármacos está por determinar93. En el canal de sodio Nav1.5 también se han documentado polimorfismos que pueden conferir susceptibilidad a desarrollar arritmias ventriculares, como sucede con el polimorfismo H558R, que se presenta hasta en un 30% de la población, o el S1103Y, frecuente en afroamericanos80,81,90,94,95 y cuya implicación en la susceptibilidad a determinados fármacos no ha sido explorada.
SINDROME DE QT LARGO Y EMBARAZO
El consejo genético es importante en esta enfermedad, pero en términos generales no hay contraindicación para el embarazo en las pacientes portadoras de SQTL, aunque cada caso en particular es diferente y debe evaluarse en su apropiado contexto.
Se ha observado que el riesgo de presentar arritmias ventriculares malignas disminuye durante la gestación. Por el contrario, hasta 9 meses después del parto se ha comunicado una mayor vulnerabilidad a presentar arritmias malignas, en especial en las pacientes portadoras de SQTL2. Este riesgo disminuye de forma importante con el tratamiento con bloqueadores beta96.
ESTRATIFICACION DEL RIESGO
La evolución en el SQTL es muy variable y está influida por la duración del intervalo QTc, los factores ambientales, la edad, el genotipo y la respuesta al tratamiento97,98. Las arritmias ventriculares son más frecuentes en SQTL1 y SQTL2, pero son más letales en SQTL399. Como mencionamos, en el posparto las mujeres son particularmente susceptibles a las arritmias malignas14.
Debe considerarse de alto riesgo el SQTL asociado con:
1. Sordera congénita (síndrome de Jervell-Lange-Nielsen).
2. Síncope recurrente por taquiarritmias ventriculares malignas.
3. Antecedentes familiares de muerte súbita.
4. QTc > 500 ms.
5. Bloqueo auriculoventricular 2:1.
6. Alternancia eléctrica en la onda T.
7. Genotipo de SQTL tipo 3.
Los estudios realizados por Priori et al97 en 647 pacientes mostraron que la probabilidad de presentar un evento mayor (síncope, paro cardiaco, muerte súbita) antes de los 40 años es alto (> 50%) cuando el QTc > 500 ms en SQTL1, SQTL2 o bien en los varones cuando existe SQTL3. Recientemente se ha comunicado el análisis del registro internacional de SQTL. Se analizó el riesgo de muerte súbita en 2.772 adolescentes con la enfermedad y se concluyó que 3 eventos se asocian con mayor riesgo en esta población: QTc > 530 ms, historia de síncope en los últimos 10 años y sexo; los varones de 10-12 años tuvieron mayor riesgo que las mujeres, pero en el rango de edad de 13-20 años, el riesgo era el mismo100.
TRATAMIENTO
Los pacientes sintomáticos que no reciben tratamiento tienen una mortalidad del 20% al año y del 50% a los 10 años después de un primer evento de arritmia ventricular. Si bien está claro que en presencia de síntomas hay que instaurar un tratamiento, la conducta que se debe seguir en los pacientes asintomáticos es aún motivo de debate. Se ha documentado que la parada cardiaca puede ser la primera manifestación de la enfermedad en el 9% de los casos48, y 12% de los casos asintomáticos desarrollará síntomas e, incluso, muerte súbita. El tratamiento inicial serán los fármacos bloqueadores beta y deben iniciarse en todo paciente con SQTL como medida inicial; la restricción en el ejercicio será recomendable pero, sin duda, los marcadores clínicos y electrocardiográficos de riesgo son de gran utilidad en la toma de decisiones. Siempre será importante informar a los pacientes sobre el riesgo de la utilización de los diversos fármacos que, como hemos mencionado con anterioridad, pueden prolongar el intervalo QT y propiciar así arritmias ventriculares.
El diagnóstico genético, además de permitir un apropiado consejo familiar en relación con la enfermedad, ayuda a evaluar el pronóstico y permite orientar de forma específica el tratamiento.
Bloqueadores beta
Constituyen la primera línea de tratamiento; todo paciente con diagnóstico de SQTL debe recibir bloqueadores beta de forma inicial101. Esta terapia reduce el riesgo de eventos hasta en un 64%100. Son eficaces, en particular, en los pacientes con mutaciones en el canal IKs (SQTL1)102, regulado de forma importante por el sistema simpático. No modifican el intervalo QT, pero sí su dispersión103.
Se ha demostrado que, si bien los bloqueadores beta disminuyen la incidencia de eventos cardiovasculares104,105, un 10% de los pacientes con SQTL1, un 23% con SQTL2 y un 32% con SQTL3 tendrán síntomas a pesar del tratamiento106. En particular, los pacientes con SQTL3 no parecen obtener un beneficio importante; de hecho, este grupo de fármacos deberá usarse con cautela, pues los episodios de arritmia ventricular en el SQTL3 son más comunes con frecuencias cardiacas bajas. En términos generales, el 32% de los pacientes sintomáticos antes del inicio del tratamiento con bloqueadores beta tendrá recurrencia de los síntomas en los primeros 5 años, y el 14% de los que fueron rescatados de un episodio de muerte súbita presentará otro evento similar en 5 años si sólo reciben esta terapia107.
Se han utilizado diversos bloqueadores beta en el tratamiento del SQTL: nadolol (0,5-1 mg/kg/día), propranolol (2-4 mg/kg/día), metoprolol (0,5-1 mg/kg/día) y atenolol (0,5-1 mg/kg/día), principalmente. Este último podría no ser beneficioso en el SQTL, pues se ha notificado que al menos el 75% de los casos que no respondieron a la terapia con bloqueadores beta recibía atenolol, aunque este hallazgo podría estar relacionado con el uso de dosis subóptimas104.
Para establecer la dosis adecuada es útil la prueba de esfuerzo. La frecuencia cardiaca máxima no debe superar los 130 lat/min en tratamiento.
Bloqueadores de los canales de sodio
Las mutaciones en el canal de sodio que ocasionan el SQTL3 producen una inactivación defectuosa del canal; los bloqueadores de los canales de sodio en estos pacientes han mostrado ser de utilidad. Estudios realizados con flecainida muestran que mejora la frecuencia cardiaca, las alteraciones en la onda T y el intervalo QT108. La mexiletina también ha mostrado mejorar los marcadores electrocardiográficos de riesgo63,109,110. Estudios in vitro con ranolazina han mostrado que disminuye los efectos deletéreos condicionados por mutaciones comunicadas en el humano111. Si bien los resultados son alentadores, hay que considerar que no hay un estudio a largo plazo que evalúe esta terapia, ni se ha comunicado en un número importante de pacientes. Los bloqueadores de los canales de sodio no se deben administrar si no hay un diagnóstico genético confirmado.
Potasio suplementario y fármacos que facilitan su disponibilidad
El suplemento de potasio y/o la utilización de fármacos ahorradores de potasio, como la espironolactona, pueden disminuir el intervalo QTc hasta en un 24% de los casos112,113. Los fármacos que favorecen la apertura de los canales de potasio, como apricalium, leveromakalium, nicornadil y pinacidil, han mostrado ser útiles en el tratamiento del SQTL. Los subtipos que en particular se benefician son el SQTL1 y el SQTL2114.
Marcapasos y desfibrilador
La estimulación con marcapasos ha sido utilizada en los pacientes con arritmia dependiente de pausa115,116. Los pacientes con un SQTL3 suelen beneficiarse más de este tratamiento, pues la prevalencia de bradicardia es mayor. Se indica estimulación DDD en los casos con arritmia dependiente de pausa o bloqueo AV 2 1 o de alto grado. Las frecuencias programadas por debajo de 70 lat/min117 no son útiles para prevenir las arritmias ventriculares. Se recomienda programar el sensor a una respuesta rápida, pues estos pacientes suelen tener una aceleración inapropiada de la frecuencia cardiaca en respuesta al ejercicio. Todas las funciones que impliquen la presencia de pausas deben ser apagadas, como la histéresis y la función nocturna. El PVARP (período refractario auricular posventricular) debe ser lo más corto posible. La función de regulación de frecuencia debe ser encendida para prevenir la pausa postextrasistólica. Hay que recordar que el sobresensado de la onda T y los fallos en la captura también pueden dar lugar a pausas.
El desfibrilador automático implantable (DAI), junto con la terapia con bloqueadores beta, disminuye de forma importante la incidencia de muerte súbita118-120. Su indicación es clara en los casos catalogados como de alto riesgo121. La programación del dispositivo variará según las necesidades del caso pero, en general, hay que evitar administrar tratamiento en los eventos autolimitados asintomáticos; para este fin, se indica un tiempo de detección > 15 s. Una complicación de la terapia con DAI es la tormenta arrítmica; se ha comunicado que cerca del 15% de los pacientes pueden presentarla y se debe, en buena parte, al incremento del tono simpático después de la descarga del DAI118; este problema puede tratarse con un incremento en la dosis de bloqueadores beta. Si esta medida no es útil se debe considerar realizar una resección de la cadena ganglionar simpática.
Simpatectomía izquierda
Desde 1971 se introdujo la gangliectomía simpática como opción terapéutica útil en estos pacientes122. En 1991, Schwartz et al123 publicaron la primera serie de 85 casos con escasa respuesta al tratamiento con bloqueadores beta, en los que se realizó estelectomía izquierda con resultados alentadores, pues se logró una supervivencia a los 5 años del 94%. Actualmente se ofrece esta opción terapéutica a pacientes de alto riesgo cuando, a pesar del tratamiento con bloqueadores beta y/o marcapasos, el paciente persiste con síncope, o bien, si una vez implantado el desfibrilador, hay descargas frecuentes.
El procedimiento consiste en realizar la resección de la porción inferior del ganglio estrellado y de las cadenas ganglionares torácicas simpáticas izquierdas T2 a T4, ya que la simple estelectomía izquierda ha mostrado no ser suficientemente efectiva. Se ha utilizado la toracoscopia microinvasiva con buenos resultados124,125. De manera reciente se ha publicado la serie más importante de pacientes tratados con este método, la cual mostró una reducción significativa en los episodios de síncope o muerte súbita, así como una supervivencia a los 5 años del 95%. En pacientes con síncope previo, la supervivencia a los 5 años fue del 97%, con un 11% de posibilidades de recurrencia, que en su mayoría consistieron en un único evento sincopal aislado. Asimismo, se encontró una reducción significativa del segmento QT posterior a la simpatectomía izquierda. A pesar de los buenos resultados, la prevención de muerte súbita no es completa, pero se logra reducir a un 3%. En pacientes portadores de un DAI en los que se realizó la cirugía por la presencia de múltiples descargas, la media de eventos fue de 25 a 0, con una reducción del 95%. En el SQTL1, se confirmó el efecto benéfico. Probablemente, el beneficio sea menor en pacientes con SQTL2. Hasta ahora no se ha logrado establecer su efectividad para el SQTL3126.
Ablación
Se ha comunicado que es posible realizar ablación de la extrasístole, que en algunos casos inicia la arritmia ventricular, con mejoría en la incidencia de episodios127. Sin embargo, no hay estudios a largo plazo ni con un número de pacientes apropiado que justifique la utilización esta técnica de forma sistemática.
Full English text available from: www.revespcardiol.org
ABREVIATURAS
AV: auriculoventricular.
DAI: desfibrilador automático implantable.
ECG: electrocardiograma.
QTc: QT corregido por frecuencia.
SAT: síndrome de Andersen-Tawil.
SQTL: síndrome de QT largo.
Véase editorial en págs. 675-82
La Dra. Medeiros recibe soporte económico de CONACyT y FUNSALUD.
Correspondencia: Dra. A. Medeiros Domingo.
Unidad de Biología Molecular. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Vasco de Quiroga, 15. Tlalpan 14000 México DF.
Correo electrónico: argeliamed@yahoo.com