ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2023 - El Congreso de la Salud Cardiovascular

Málaga,
26 - 29 de Octubre de 2023
Introducción
Dr. Juan José Gómez Doblas
Presidente del Comité Científico del Congreso
Vicepresidente de la SEC
Comités ejecutivo, organizador y científico
Comité de evaluadores
Listado de sesiones
Índice de autores
89. Nuevas aplicaciones de la genética en el área cardiovascular
Fecha
: 28-10-2023 10:15:00
Tipo
: Comunicaciones mini orales
Sala
: Sala M5
7. REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
Ana I. Fernández Ávila1, María Ángeles Espinosa Castro2, Irene Méndez Fernández2, Silvia Vilches Soria2, Cristina Gómez García2, Reyes Álvarez García-Rovés3, Miriam Centeno Jiménez3, María López Blázquez3, Nélida Vázquez Aguilera2, Constancio Medrano López3, Javier Bermejo Thomas2 y Francisco Fernández-Avilés Díaz2
1Instituto de Investigación Sanitaria Gregorio Marañón. Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares CIBER-CV, Madrid, España, 2Programa CSUR de Cardiopatías Familiares. Servicio de Cardiología de Adultos. Hospital General Universitario Gregorio Marañón, Madrid, España y 3Programa CSUR de Cardiopatías Familiares. Servicio de Cardiología Infantil. Hospital General Universitario Gregorio Marañón, Madrid, España.
1Instituto de Investigación Sanitaria Gregorio Marañón. Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares CIBER-CV, Madrid, España, 2Programa CSUR de Cardiopatías Familiares. Servicio de Cardiología de Adultos. Hospital General Universitario Gregorio Marañón, Madrid, España y 3Programa CSUR de Cardiopatías Familiares. Servicio de Cardiología Infantil. Hospital General Universitario Gregorio Marañón, Madrid, España.
Introducción y objetivos: Las variantes genéticas causales en miocardiopatía dilatada (MCD) siguen siendo en gran parte desconocidas. Una proporción de la heredabilidad faltante podría localizarse en regiones genómicas inexploradas. Los análisis de asociación de genomas completos (GWAS) permiten el descubrimiento de nuevos loci. Estudios GWAS recientes revelan que existen regiones adicionales que incluyen genes candidato funcionales relevantes a considerar en MCD (figura). Objetivo: revisión de variantes en los genes candidato propuestos a partir de los últimos análisis GWAS en MCD (NEDD4L, MAP3K7CL, CDKN1A, HSPB7, SMARCB1 y SLC6A6) como potenciales causales en una cohorte de pacientes con cardiopatías familiares.
Métodos: Se revisaron retrospectivamente los análisis genéticos (Exoma clínico o completo) de 896 pacientes índices de un programa de Cardiopatías Familiares de un centro de referencia entre marzo de 2019 y agosto de 2022. Analizamos el fenotipo del paciente, tipo de variante y patrones de variabilidad de los genes descritos previamente, tomando como referencia el gen causal MYH7.
Resultados: En ningún gen se identificaron variantes truncantes a excepción del gen SLC6A6, donde se observó un enriquecimiento de estas (91% pacientes), descartando su causalidad (figura). Diez variantes missense en 11 pacientes cumplían criterios -ausencia en población general, potencial efecto deletéreo y sin otra variante causal- para ser consideradas candidatas a analizar en estudios de segregación familiar (tabla). Especialmente interesante resultó el gen NEDD4L. Cinco pacientes, 2 con MCD y 3 con canalopatías portaban variantes candidatas. Dos de ellos familiares de primer grado con una melladura similar en QT con la bipedestación. El gen NEDD4L codifica para una ligasa que participa en la degradación de canales de sodio. Modificaciones de la interacción SCNA5-NEDD4L representan un nuevo mecanismo de patogenicidad para las enfermedades relacionadas con alteración en los canales NaV1.5, incluyendo síndrome de Brugada, QT largo y MCD.
|
Variantes missense candidatas (MAF< 0,01%, Predicción deletérea) a analizar en estudios de segregación familiar |
|||||||||||
|
Paciente |
Fenotipo principal |
Gene |
Transcrito |
cDNA |
Fracción |
Proteína |
Predicción impacto |
dbSnpID |
ClinVar |
MAF población |
Identificada variante causal |
|
1 |
SB |
NEDD4L |
NM_001144964 |
c.712C>T |
0,5 |
p.(Pro238Ser) |
Potential alteration auxiliary splicing |
|
|
0,000% |
No |
|
2 |
SQTL* |
NEDD4L |
NM_001144967 |
c.331C>A |
0,5 |
p.(Pro111Thr) |
Activation of a cryptic Donor site |
|
|
0,000% |
SCN5A |
|
3 |
SQTL* |
NEDD4L |
NM_001144967 |
c.331C>A |
0,5 |
p.(Pro111Thr) |
Activation of a cryptic Donor site |
|
|
0,000% |
No |
|
4 |
MCD |
NEDD4L |
NM_001144964 |
c.860A>G |
0,5 |
p.(Asn287Ser) |
Create or strengthen a splice site |
rs759746029 |
VUS |
0,000% |
No |
|
5 |
MCD |
NEDD4L |
NM_001144968 |
c.7C>T |
0,5 |
p.(Arg3Cys) |
Potential alteration auxiliary splicing |
rs746580879 |
|
0,001% |
No |
|
6 |
MCD |
CDKN1A |
NM_000389 |
c.136C>T |
0,5 |
p.(Arg46Cys) |
Untolerated change |
rs762978904 |
|
0,000% |
No |
|
7 |
MCD |
HSPB7 |
NM_014424 |
c.478G>A |
0,5 |
p.(Val160Ile) |
Untolerated change |
rs144366876 |
|
0,009% |
No |
|
8 |
MCD |
HSPB7 |
NM_014424 |
c.470C>T |
0,5 |
p.(Thr157Ile) |
Untolerated change |
rs769371992 |
|
0,000% |
No |
|
9 |
MCD |
HSPB7 |
NM_014424 |
c.485A>C |
0,5 |
p.(Gln162Pro) |
Untolerated change |
rs142826385 |
|
0,000% |
No |
|
10 |
MCD |
SMARCB1 |
NM_003073 |
c.735C>G |
0,5 |
p.(Ile245Met) |
Untolerated change |
|
|
0,000% |
No |
|
11 |
MCD |
SLC6A6 |
NM_001134367 |
c.646A>C |
0,5 |
p.(Lys216Gln) |
Untolerated change |
|
|
0,000% |
No |
|
*Familiares de primer grado con melladura en QT con la bipedestación. |
|||||||||||
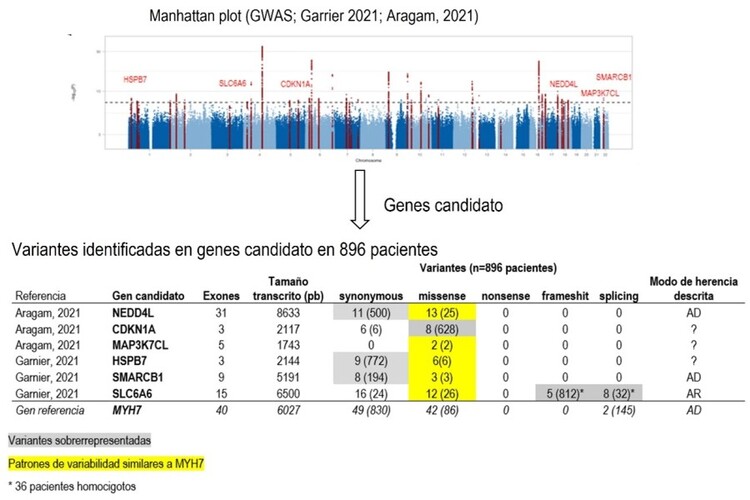
Variantes identificadas, tipo y frecuencia, para los genes candidatos analizados.
Conclusiones: El análisis de nuevos genes candidato funcionales a partir de resultados de GWAS ha permitido identificar variantes relevantes, especialmente en el gen NEDD4L, que deben ser analizadas en mayor profundidad.
Comunicaciones disponibles de "Nuevas aplicaciones de la genética en el área cardiovascular"
- 1. MODERA
- Alejando Isidoro Pérez Cabeza, Hospital Universitario Virgen de la Victoria, Málaga
- 2. HETEROGENEIDAD GENÉTICA EN EL ANEURISMA DE AORTA ASCENDENTE: IDENTIFICACIÓN DIFERENCIAL DE NUEVAS DIANAS TERAPÉUTICAS
- Antonio José Barros Membrilla1, Álvaro Rodríguez Pérez1, Rafael Almendra Pegueros2, Juan Francisco Tabilo Ahumada3, Laura Martín-Fernández4, Elvira Pérez Marlasca5, Francisco Vidal6, José Martínez González7, Cristina Rodríguez Sinovas2 y María Galán Arroyo5
1Cardiología. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 2Institut de Recerca IIB-Sant Pau, Barcelona, España, 3Cirugía Cardiaca. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 4Laboratorio de Coagulopatías congénitas. Banco de Sangre y Tejidos BST, Barcelona, España, 5Ciencias de la Salud. Universidad Rey Juan Carlos, Alcorcón Madrid, España, 6Laboratorio de Coagulopatías Congénitas. Banco de Sangre y Tejidos BST, Barcelona, España y 7Institut d'Investigacions Biomèdiques de Barcelona.IIBB-CSIC., Barcelona, España.
- 3. FIBRAS DE PURKINJE EN LA TAQUICARDIA VENTRICULAR POLIMÓRFICA CATECOLAMINÉRGICA
- Sara Huélamo Montoro1, Jorge Manuel Sanz Ros1, Carlos Fernández Sellers2, Amparo Ruiz Sauri3, Cinta Moro4, Cristina Presentación2, Jennifer Sancho Jiménez2, Rafael Sánchez5, Marta Sepúlveda5, Pilar Molina Aguilar2, Javier Navarrete Navarro6 y Esther Zorio Grima7
1Unidad de Cardiopatías Familiares. Servicio de Cardiología. Hospital Universitari i Politècnic La Fe, Valencia, España, 2Servicio de Patología. Instituto de Medicina Legal y Forense, Valencia. CAFAMUSME, Grupo acreditado de Cardiopatías Familiares, Muerte Súbita y Mecanismos de Enfermedad. Instituto de Investigación Sanitaria La Fe IIS La Fe, Valencia, España, 3Departamento de Patología. Facultad de Medicina y Odontología, Universidad de Valencia, Valencia, España, 4Servicio de Histopatología. Instituto Nacional de Toxicología y Ciencias Forenses, Sevilla, España, 5CAFAMUSME, Grupo Acreditado de Cardiopatías Familiares, Muerte Súbita y Mecanismos de Enfermedad. Instituto de Investigación Sanitaria La Fe IIS La Fe, Valencia, España, 6Servicio de Cardiología. Hospital Universitari i Politècnic La Fe, Valencia, España y 7CAFAMUSME, Grupo Acreditado de Cardiopatías Familiares, Muerte Súbita y Mecanismos de Enfermedad. Unidad de Cardiopatías Familiares, Servicio de Cardiología. Hospital Universitari i Politècnic La Fe. CIBERCV, Valencia, España.
- 4. CARACTERIZACIÓN ELECTROFISIOLÓGICA DE LAS TAQUICARDIAS VENTRICULARES MONOMÓRFICAS SOSTENIDAS EN PACIENTES CON MIOCARDIOPATÍA ARRITMOGÉNICA IZQUIERDA: ESTUDIO DE CORRELACIÓN GENOTIPO-FENOTIPO
- Eva Cabrera Borrego, Francisco José Bermúdez Jiménez, Rosa Macías Ruíz, Pablo J. Sánchez Millán y Juan Jiménez Jáimez
Cardiología. Hospital Universitario Virgen de las Nieves. Instituto de Investigación Biosanitaria ibs.Granada, Granada, España.
- 5. EL PEZ CEBRA MUTANTE DSPB-/- TERT+/- COMO MODELO DE MIOCARDIOPATÍA ARRITMOGÉNICA: EL IMPACTO DEL ENVEJECIMIENTO EN EL DESARROLLO DE LA ENFERMEDAD
- Serena Evelina Margaretha Munteanu1, Jesús Wagih Gómez2, Elisa Nicolás Rocamora3, Cristina Gil Ortuño3, Ángel Bernabé García4, Eva Cabrera-Romero5, María Luisa Cayuela Fuentes6, Juan Ramón Gimeno Blanes7 y María Sabater Molina8
1Cardiogenética. Instituto Murciano de Investigación Biosanitaria IMIB Pascual Parrilla, Murcia, España, 2Cardiogenética. Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia, España, 3Universidad de Murcia, Murcia, España, 4Instituto Murciano de Investigación Biosanitaria IMIB Pascual Parrilla, Murcia, España, 5Unidad de Cardiopatías Familiares, Servicio de Cardiología. Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), España, 6Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia, España, 7Unidad de Cardiopatías Familiares, Servicio de Cardiología. Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, España y 8Instituto Murciano de Investigación Biosanitaria, Cardiogenética. Universidad de Murcia, Murcia, España.
- 6. EL PEZ CEBRA MUTANTE DSPB-/- COMO MODELO PARA LA MIOCARDIOPATÍA ARRITMOGÉNICA: EL IMPACTO DE LA ACTIVIDAD FÍSICA
- Serena Evelina Margaretha Munteanu1, Jesús Wagih Gómez1, Elisa Nicolás Rocamora2, Cristina Gil Ortuño2, Ángel Bernabé García3, Eva Cabrera-Romero4, María Luisa Cayuela Fuentes5, Juan Ramón Gimeno Blanes6 y María Sabater Molina7
1Cardiogenética. Instituto Murciano de Investigación Biosanitaria IMIB Pascual Parrilla, Murcia, España, 2Universidad de Murcia, Murcia, España, 3Instituto Murciano de Investigación Biosanitaria IMIB Pascual Parrilla, Murcia, España, 4Unidad de Cardiopatías Familiares, Servicio de Cardiología. Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), España, 5Cirurgía Digestiva, Endocrina y Trasplante de Órganos Abdominales, Área de Cirugía Experimental. Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia, España, 6Unidad de Cardiopatías Familiares, Servicio de Cardiología. Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, España y 7Cardiogenética, Instituto Murciano de Investigación Biosanitaria. Universidad de Murcia, Murcia, España.
- 7. REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- Ana I. Fernández Ávila1, María Ángeles Espinosa Castro2, Irene Méndez Fernández2, Silvia Vilches Soria2, Cristina Gómez García2, Reyes Álvarez García-Rovés3, Miriam Centeno Jiménez3, María López Blázquez3, Nélida Vázquez Aguilera2, Constancio Medrano López3, Javier Bermejo Thomas2 y Francisco Fernández-Avilés Díaz2
1Instituto de Investigación Sanitaria Gregorio Marañón. Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares CIBER-CV, Madrid, España, 2Programa CSUR de Cardiopatías Familiares. Servicio de Cardiología de Adultos. Hospital General Universitario Gregorio Marañón, Madrid, España y 3Programa CSUR de Cardiopatías Familiares. Servicio de Cardiología Infantil. Hospital General Universitario Gregorio Marañón, Madrid, España.
- 8. IDENTIFICACIÓN DE NUEVOS LOCI IMPLICADOS EN LA DISFUNCIÓN CARDIACA RELACIONADA CON EL TRATAMIENTO DEL CÁNCER MEDIANTE EL METANÁLISIS DE ESTUDIOS DE ASOCIACIÓN DE GENOMA COMPLETO
- Laura Martínez Campelo1, Alejandro Blanco Verea1, Teresa López Fernández2, Amparo Martínez Monzonís3, Antonio Buño Soto4, Pilar Mazón Ramos3, Nadine Norton5, Alejandro Velasco6, Ángel Carracedo Álvarez7, Jose Ramón González-Juanatey8, José Luis López-Sendón Hentschel9 y María Brion10
1Genética Cardiovascular. Instituto de Investigación Sanitaria Santiago de Compostela IDIS, Santiago de Compostela (A Coruña), España, 2Servicio de Cardiología. Hospital Universitario La Paz, Madrid, España, 3Servicio de Cardiología. Hospital Clínico Universitario de Santiago de Compostela, Investigación Sanitaria de Santiago de Compostela IDIS y CIBERCV, Santiago de Compostela (A Coruña), España, 4Servicio de Análisis Clínicos. Hospital Universitario La Paz, Madrid, España, 5Department of Cancer Biology. Mayo Clinic, Jacksonville Florida, Estados Unidos, 6Centro Nacional de Investigaciones Oncológicas CNIO, Madrid, España, 7Medicina Xenómica, Universidade de Santiago de Compostela. CIBERER. Fundación Pública Galega de Medicina Xenómica, Santiago de Compostela (A Coruña), España, 8Servicio de Cardiología. Hospital Clínico Universitario de Santiago, Instituto de Investigación Sanitaria de Santiago IDIS, CIBER-CV, Santiago de Compostela (A Coruña), España, 9Instituto de Investigación Hospital Universitario La Paz, Madrid, España y 10Hospital Clínico Universitario de Santiago, Instituto de Investigación Sanitaria de Santiago IDIS, CIBER-CV, Santiago de Compostela (A Coruña), España.
Más comunicaciones de los autores
-
Álvarez García-Rovés, Reyes
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
-
Bermejo Thomas, Javier
- 7 - EL EFECTO TERAPÉUTICO DE VESÍCULAS EXTRACELULARES EMBEBIDAS EN HIDROGEL DE MATRIZ CARDIACA EN MODELO PORCINO DE CARDIOPATÍA ISQUÉMICA
- 5 - ¿CUÁL ES LA MEJOR ESTRATEGIA DE DESCARGA EN PACIENTES CON ECMO?
- 13 - CORRELACIÓN DE LA CLASE FUNCIONAL OBJETIVA CON PARÁMETROS HEMODINÁMICOS NO INVASIVOS EN PACIENTES CON ANOMALÍA DE EBSTEIN
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
- 7 - TRATAMIENTO DEL EMBOLISMO PULMONAR CON TROMBECTOMÍA PERCUTÁNEA: EXPERIENCIA DE UN CENTRO
- 11 - TRATAMIENTO DE LAS ARRITMIAS VENTRICULARES REFRACTARIAS MEDIANTE DENERVACIÓN SIMPÁTICA CARDIACA QUIRÚRGICA
- 5 - ¿ES SEGURO EL SOPORTE CIRCULATORIO PROLONGADO CON IMPELLA? EXPERIENCIA EN UNA COHORTE RETROSPECTIVA DE SHOCK CARDIOGÉNICO EN UN CENTRO DE TERCER NIVEL
- 13 - CORRESPONDENCIA DE LOS CANALES DE CONDUCCIÓN LENTA IDENTIFICADOS EN LOS MAPAS DE VOLTAJE DURANTE UNA TAQUICARDIA VENTRICULAR RÁPIDA CON AQUELLOS IDENTIFICADOS EN LOS MAPAS DE VOLTAJE EN SINUSAL
- 8 - DIFERENCIAS EN ERGOESPIROMETRÍA EN CICLOERGÓMETRO SUPINO DURANTE CATETERISMO DE ESFUERZO FRENTE A TAPIZ RODANTE CONVENCIONAL EN PACIENTES CON CIRCULACIÓN DE FONTAN
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
-
Centeno Jiménez, Miriam
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
-
Espinosa Castro, María Ángeles
- 12 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA PEDIÁTRICA POR VARIANTES EN MYH7
- 17 - RESULTADOS PRELIMINARES DEL REGISTRO ESPAÑOL DE AMILOIDOSIS HEREDITARIA POR TRANSTIRRETINA
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
- 5 - IMPACTO DEL GENOTIPO EN LA RESPUESTA A TRATAMIENTO MÉDICO A LARGO PLAZO EN MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
-
Fernández Ávila, Ana I.
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
-
Fernández-Avilés Díaz, Francisco
- 13 - CORRELACIÓN DE LA CLASE FUNCIONAL OBJETIVA CON PARÁMETROS HEMODINÁMICOS NO INVASIVOS EN PACIENTES CON ANOMALÍA DE EBSTEIN
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
- 11 - CARACTERIZACIÓN HÍBRIDA EN FIBRILACIÓN AURICULAR Y RITMO SINUSAL DURANTE MAPEO ELECTROANATÓMICO DE ULTRA ALTA DENSIDAD
- 11 - TRATAMIENTO DE LAS ARRITMIAS VENTRICULARES REFRACTARIAS MEDIANTE DENERVACIÓN SIMPÁTICA CARDIACA QUIRÚRGICA
- 13 - CORRESPONDENCIA DE LOS CANALES DE CONDUCCIÓN LENTA IDENTIFICADOS EN LOS MAPAS DE VOLTAJE DURANTE UNA TAQUICARDIA VENTRICULAR RÁPIDA CON AQUELLOS IDENTIFICADOS EN LOS MAPAS DE VOLTAJE EN SINUSAL
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 8 - DIFERENCIAS EN ERGOESPIROMETRÍA EN CICLOERGÓMETRO SUPINO DURANTE CATETERISMO DE ESFUERZO FRENTE A TAPIZ RODANTE CONVENCIONAL EN PACIENTES CON CIRCULACIÓN DE FONTAN
- 5 - ¿ES SEGURO EL SOPORTE CIRCULATORIO PROLONGADO CON IMPELLA? EXPERIENCIA EN UNA COHORTE RETROSPECTIVA DE SHOCK CARDIOGÉNICO EN UN CENTRO DE TERCER NIVEL
- 2 - USO DE PRÓTESIS BALÓN EXPANDIBLE MYVAL EN INSUFICIENCIA AÓRTICA NO CALCIFICADA. RESULTADOS A CORTO Y LARGO PLAZO
- 7 - TRATAMIENTO DEL EMBOLISMO PULMONAR CON TROMBECTOMÍA PERCUTÁNEA: EXPERIENCIA DE UN CENTRO
- 6 - SEGURIDAD Y EFICACIA DE LOS AGONISTAS DEL RECEPTOR DE PÉPTIDO SIMILAR AL GLUCAGÓN TIPO 1 EN TRASPLANTE CARDIACO
- 7 - EL EFECTO TERAPÉUTICO DE VESÍCULAS EXTRACELULARES EMBEBIDAS EN HIDROGEL DE MATRIZ CARDIACA EN MODELO PORCINO DE CARDIOPATÍA ISQUÉMICA
- 3 - PAPEL DE LA QUINIDINA EN LA TAQUICARDIA VENTRICULAR POLIMÓRFICA CON QT NORMAL DESENCADENADA POR EXTRASISTOLIA VENTRICULAR DE ACOPLAMIENTO CORTO: EXPERIENCIA DE UN CENTRO
- 2 - RESULTADOS A MEDIO PLAZO DEL REEMPLAZO VALVULAR AÓRTICO EN PACIENTES INGRESADOS E INESTABLES. EXPERIENCIA DE UN CENTRO
- 9 - RESULTADOS DE LA INTERVENCIÓN CORONARIA PERCUTÁNEA EN LOS PACIENTES CON DISECCIÓN CORONARIA ESPONTÁNEA
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
- 5 - ¿CUÁL ES LA MEJOR ESTRATEGIA DE DESCARGA EN PACIENTES CON ECMO?
- 6 - IMPACTO DEL BLOQUEO DEL SISTEMA RENINA-ANGIOTENSINA EN LA EVOLUCIÓN CLÍNICA Y EL REMODELADO VENTRICULAR IZQUIERDO TRAS IMPLANTE DE PRÓTESIS AÓRTICA TRANSCATÉTER: ESTUDIO ALEATORIZADO RASTAVI
- 7 - REVERSIBILIDAD DEL DETERIORO COGNITIVO EN PACIENTES CON INSUFICIENCIA CARDIACA AVANZADA TRAS EL TRASPLANTE CARDIACO
- 2 - ESTUDIO DE LA DOSIS ÓPTIMA DE VESÍCULAS EXTRACELULARES DERIVADAS DE CARDIOESFERAS PARA EFECTO ANTISENESCENCIA
- Gómez García, Cristina
- López Blázquez, María
-
Medrano López, Constancio
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
-
Méndez Fernández, Irene
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
- 10 - EVALUACIÓN SISTEMÁTICA PROSPECTIVA DE PACIENTES JÓVENES CON IMPLANTACIÓN DE MARCAPASOS IDIOPÁTICA
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
-
Vázquez Aguilera, Nélida
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
-
Vilches Soria, Silvia
- 4 - MUERTE SÚBITA Y CARDIOPATÍA ASOCIADA CON EL GEN PPA2. DESCRIPCIÓN FENOTÍPICA Y GENÉTICA DE LA PRIMERA FAMILIA AFECTADA E IDENTIFICADA EN ESPAÑA CON ESTA RARA Y GRAVE PATOLOGÍA
- 7 - REVISIÓN DE VARIANTES EN NUEVAS REGIONES GENÓMICAS ASOCIADAS A MIOCARDIOPATÍA DILATADA EN UNA COHORTE DE PACIENTES CON CARDIOPATÍAS HEREDITARIAS
- 9 - DIAGNÓSTICO DE MIOCARDIOPATÍA ARRITMOGÉNICA TRAS INFECCIÓN POR SARS-COV-2: SERIE DE CASOS