ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2024 - El Congreso de la Salud Cardiovascular

Bilbao,
24 - 26 de Octubre de 2024
Introducción
Dr. José María de la Torre Hernández
Presidente del Comité Científico del Congreso. Vicepresidente de la SEC
Comités ejecutivo, organizador y científico
Comité de evaluadores
Listado completo de comunicaciones
Índice de autores
5002. Miscelánea en cardiogenética y cardiopatías familiares
Fecha
: 24-10-2024 09:00:00
Tipo
: Comunicaciones mini orales
Moderadores
: Ainhoa Robles Mezcua, Hospital Universitario Virgen de la Victoria, Málaga. IBIMA. CIBER-CV
5002-3. Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
David Belmar Clivillé1, Carlos Moliner Abós1, Benjamín Rodríguez Santiago2, Javier Limeres Freire3, Clara Badia Molins3, Patricia Muñoz Cabello4, Jara Gayán Ordás5, Fernando de Frutos Seminario6, Clara Serra Juhè2, Alicia Artigas Baleri2, Marta de Antonio Ferrer1, Marta Campreciós Crespo1 y Sonia Mirabet Pérez1
1Cardiología. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 2Genética. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 3Cardiología. Hospital Universitari Vall d'Hebron, Barcelona, España, 4Genética. Hospital Universitari Vall d'Hebron, Barcelona, España, 5Cardiología. Hospital Universitari Arnau de Vilanova, Lleida, España y 6Cardiología. Hospital Bellvitge, L'Hospitalet de Llobregat (Barcelona), España.
1Cardiología. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 2Genética. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 3Cardiología. Hospital Universitari Vall d'Hebron, Barcelona, España, 4Genética. Hospital Universitari Vall d'Hebron, Barcelona, España, 5Cardiología. Hospital Universitari Arnau de Vilanova, Lleida, España y 6Cardiología. Hospital Bellvitge, L'Hospitalet de Llobregat (Barcelona), España.
Introducción y objetivos: Se han establecido correlaciones genotipo-fenotipo en la miocardiopatía hipertrófica (MCH) asociadas a un peor pronóstico (p.ej. región conversora del gen MYH7). El estudio genético realizado en múltiples pacientes con MCH identificó la variante p.(Gln892Lys) en el gen MYH7, clasificada como variante de significado incierto según las guías ACMG. Nuestro objetivo es demostrar la patogenicidad de dicha variante, y caracterizar clínicamente a los portadores estableciendo su penetrancia, pronóstico y su potencial efecto fundador en Cataluña.
Métodos: Estudio multicéntrico de cohortes retrospectivas de individuos con la variante p.(Gln892Lys) en heterocigosis en MYH7. Análisis de datos clínicos en el momento del diagnóstico y resultados clínicos durante el seguimiento. Determinación de haplotipos para valorar si existe un efecto fundador de la variante en Cataluña.
Resultados: En total, se identificaron 54 individuos portadores de la variante p.(Gln892Lys) en MYH7 (12 casos índice, 33 familiares estudiados y 9 portadores obligados). De los 45 individuos con caracterización fenotípica, 30 (67%) presentaron MCH, 1 (2%) MCD y 1 (2%) MCR. La edad media a la que se diagnosticó la miocardiopatía fue 36,1 (± 18,6) años. La clínica más frecuente al diagnóstico fue disnea en 8 (18%) pacientes. En el seguimiento, 12 (22%) pacientes fallecieron: 3 (6%) súbitamente (MS), 1 (2%) presentó una MS recuperada, 3 (6%) requirieron trasplante cardiaco y 6 (11%) implante de marcapasos. Se identificó una penetrancia completa (100%) a los 80 años, sin diferencias según género (HR 0,80, IC95% 0,43-1,48). Se hizo un estudio de haplotipos (datos de 10 casos índices) que mostró un posible origen común de la variante (efecto fundador).
|
Características basales (1ª visita): probandos vs familiares |
|||
|
|
Probandos (n = 12) |
Familiares (n = 33) |
p |
|
Edad 1ª visita (años) |
37,8 (17,6) |
39,0 (22,3) |
0,86 |
|
Mujeres |
5 (41) |
15 (45) |
0,82 |
|
Motivo 1ª visita |
< 0,01* |
||
|
Síntomas |
7 (58) |
3 (9) |
|
|
Antec. familiares |
2 (17) |
27 (84) |
|
|
Alteración ECG/soplo |
3 (25) |
2 (6) |
|
|
Clínica 1ª visita |
|||
|
Disnea |
5 (42) |
3 (9) |
0,01* |
|
Síncope |
2 (17) |
0 (0) |
0,02* |
|
Dolor torácico |
1 (8) |
1 (3) |
0,46 |
|
Palpitaciones |
5 (42) |
5 (16) |
0,07 |
|
ECG basal |
< 0,01* |
||
|
Rimo sinusal |
7 (58) |
30/30 (100) |
|
|
FA |
2 (17) |
0 |
|
|
Ritmo estimulado |
3 (25) |
0 |
|
|
Fenotipo basal |
0,03* |
||
|
Normal |
0 (0) |
12 (38) |
|
|
MCH |
11 (92) |
19 (59) |
|
|
MCD |
1 (8) |
0 |
|
|
MCR |
0 (0) |
1 (3) |
|
|
Grosor septal (mm) |
18,5 (4,0) |
13,6 (4,0) |
< 0,01* |
|
Patrón HVI |
|||
|
No HVI |
1 (9) |
12 (40) |
0,08 |
|
Septal asimétrico |
8 (73) |
17 (57) |
|
|
Concéntrico |
2 (18) |
1 (3) |
|
|
Apical |
0 (0) |
0 (0) |
|
|
Gradiente obstructivo |
|||
|
No |
10 (90) |
26 (90) |
0,81 |
|
Basal |
1 (9) |
2 (7) |
|
|
Pos-Valsalva |
0 (0) |
1 (3) |
|
|
RM cardiaca (n = 18) |
|||
|
Grosor máximo (mm) |
18,2 (2,6) |
14,8 (5,3) |
0,16 |
|
RTG |
6/6 (100) |
6/12 (50) |
0,03* |
|
ECG: electrocardiograma; FA: fibrilación auricular; MCH: miocardiopatía hipertrófica; MCD: miocardiopatía dilatada; MCR: miocardiopatía restrictiva; HVI: hipertrofia ventricular izquierda; RM: resonancia magnética; RTG: realce tardío de gadolinio. |
|||
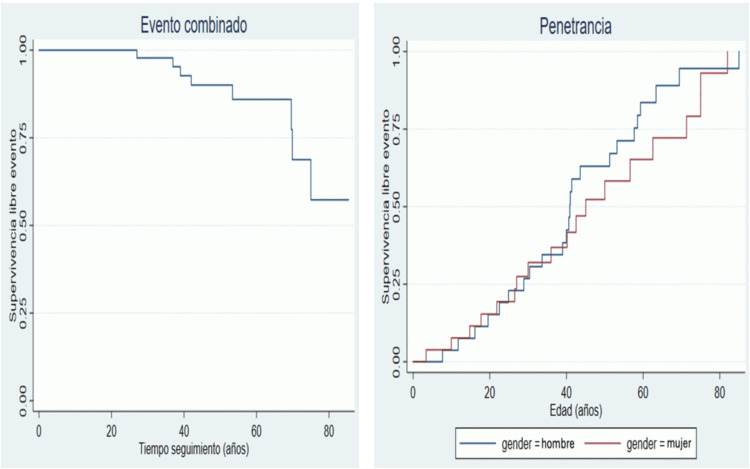
Curvas de supervivencia.
Conclusiones: La cosegregación de la variante p.(Gln892Lys) en el gen MYH7 permite reclasificarla como probablemente patogénica; con un efecto fundador en Cataluña. Esta variante es responsable de una forma de MCH con una elevada penetrancia y expresividad variable, caracterizada por una alta incidencia de trastornos de conducción que requiere implante de marcapasos.
Comunicaciones disponibles de "5002. Miscelánea en cardiogenética y cardiopatías familiares"
- 5002-1. Modera
- Ainhoa Robles Mezcua, Hospital Universitario Virgen de la Victoria, Málaga. IBIMA. CIBER-CV
- 5002-2. Valor diagnóstico y pronóstico de la cuantificación de la grasa mediante tomografía computarizada en la miocardiopatía arritmogénica del ventrículo derecho
- Valentina Faga1, María Ruiz Cueto1, David Vilades Medel2, Zoraida L. Moreno Weidmann2, Paolo Dallaglio1, Carles Díez López1, Gerard Roura Ferrer1, José M. Guerra2, Rubén Leca Petracca2, Joan Antoni Gómez Hospital1, Josep Comín Colet1, Ignasi Anguera Camos1 y Andrea di Marco1
1Hospital Universitari de Bellvitge, L'Hospitalet de Llobregat (Barcelona), España y 2Hospital de la Santa Creu i Sant Pau, Barcelona, España.
- 5002-3. Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
- David Belmar Clivillé1, Carlos Moliner Abós1, Benjamín Rodríguez Santiago2, Javier Limeres Freire3, Clara Badia Molins3, Patricia Muñoz Cabello4, Jara Gayán Ordás5, Fernando de Frutos Seminario6, Clara Serra Juhè2, Alicia Artigas Baleri2, Marta de Antonio Ferrer1, Marta Campreciós Crespo1 y Sonia Mirabet Pérez1
1Cardiología. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 2Genética. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 3Cardiología. Hospital Universitari Vall d'Hebron, Barcelona, España, 4Genética. Hospital Universitari Vall d'Hebron, Barcelona, España, 5Cardiología. Hospital Universitari Arnau de Vilanova, Lleida, España y 6Cardiología. Hospital Bellvitge, L'Hospitalet de Llobregat (Barcelona), España.
- 5002-4. Variantes missense en FLNC y miocardiopatía hipertrófica: el todo no es la suma de las partes
- María Valverde Gómez, Luis de la Higuera Romero, Soledad García Hernández, Marlene Pérez Barbeito, María Sánchez Flores, Martín Ortiz Genga, Laura Cazón Varela, Iria Gómez Díaz, Rosalía Peteiro Deben, Diego Cabrera Argaña, María Noel Brögger, Xusto Fernández Fernández, Ivonne Cárdenas Reyes, Almudena Amor Salamanca y Juan Pablo Ochoa Folmer
Cardiología. Health in Code, A Coruña, España.
- 5002-5. Rentabilidad del estudio genético en la miocardiopatía hipertrófica en una unidad especializada
- Lidia Rodero Barcos, Helena Llamas Gómez, Pablo Martín Marín, David Grimaldos Parra, Miguel Barranco Gutiérrez, María Esperanza Donoso y María Luisa Peña Peña
Hospital Universitario Virgen del Rocío, Sevilla, España.
- 5002-6. El strain de la aurícula izquierda predice eventos cardiovasculares adversos en la miocardiopatía hipertrófica: resultados preliminares de un estudio de RMC
- Francisco González Santorun1, Guillem Casas Masnou1, Andrea Faggiano2, Mateo Brusamolino3, Filipa Xavier Valente1, Ruper Oliveró Soldevila1, Clara Badia Molins1, Javier Limeres Freire1, Gisela Teixido Tura1, Eduardo Ródenas Alesina1, Andrea Guala4, Ignacio Ferreira González1 y José Fernando Rodríguez Palomares1
1Servicio de Cardiología. Hospital Universitari Vall d'Hebron, Barcelona, España, 2Departamento de Enfermedades Vasculares Cardio-Torácicas. Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano (Italia), 3Departamento de Cardiología Perioperatoria e Imagen Cardiovascular. IRCCS Centro Cardiologico Monzino, Milano (Italia) y 4Grupo de Enfermedades Cardiovasculares. Vall d'Hebron Institut de Recerca (VHIR), Barcelona, España.
- 5002-7. Genotipos de alto riesgo en miocardiopatía dilatada: más allá de las arritmias
- Nerea Mora Ayestarán1, Juan Pablo Ochoa1, M. Ángeles Espinosa Castro2, Marina Navarro Peñalver3, María Gallego Delgado4, Roberto Barriales Villa5, Ainhoa Robles Mezcua6, Javier Limeres Freire7, Mayte Basurte Elorz8, Vicente Climent Payá9, María Victoria Mogollón Jiménez10, Juan Jiménez Jáimez11, Pablo Elpidio García Granja12, Fernando Domínguez Rodríguez1 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), España, 2Hospital General Universitario Gregorio Marañón, Madrid, España, 3Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, España, 4Complejo Asistencial Universitario de Salamanca, Salamanca, España, 5Complexo Hospitalario Universitario A Coruña, A Coruña, España, 6Hospital Clínico Universitario Virgen de la Victoria, Málaga, España, 7Hospital Universitario Vall d'Hebron, Barcelona, España, 8Hospital Universitario de Navarra, Pamplona (Navarra), España, 9Hospital General Universitario de Alicante, Alicante, España, 10Hospital San Pedro de Alcántara, Cáceres, España, 11Hospital Universitario Virgen de las Nieves, Granada, España y 12Hospital Clínico Universitario de Valladolid, Valladolid, España.
- 5002-8. Evaluación de la capacidad funcional y parámetros cardiovasculares mediante CPET en pacientes con síndrome de Marfan
- Clara Badia Molins, Jordi Lozano Torres, Patricia Launois, Alba Gómez Garrido, Raúl Aguilar López, Rubén Fernández Galera, Andrea Guala, Axel Hiram Hernández, Rodrigo Fernández, Laura Galian Gay, Ruper Oliveró Soldevila, Guillem Casas Masnou, José Fernando Rodríguez Palomares y Gisela Teixido Tura
Hospital Universitari Vall d'Hebron, Barcelona, España.
- 5002-9. Impacto clínico del estudio y consejo genético en familiares de pacientes con amiloidosis TTR hereditaria
- Nerea Mora Ayestarán1, Celia Gil Llopis2, Clea González Maniega1, María Victoria Piovano1, Daniel Águila Gordo3, Belén Peiró Aventín1, Daniel de Castro Campos1, Esther González López1, Fernando Domínguez Rodríguez1 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), España, 2Hospital Universitario Dr. Peset, Valencia, España y 3Hospital General Universitario de Ciudad Real, Ciudad Real, España.
- 5002-10. Nueva estrategia de cribado de pacientes con hipercolesterolemia familiar partiendo de analítica centralizada preexistente. Estudio de las alteraciones genéticas documentadas, evolución del perfil lipídico y valoración del riesgo cardiovascular
- Joaquín Sánchez-Prieto Castillo1, Joan Ramón Enseñat1, Charlotte Boillot1, Lucía Villafáfila Martínez1, José Manuel Martínez Palomares1, Alejandro Cabello Rodríguez1, Natalia Navarro Pelegrini1, Ainhoa Aguinaga Mendibil1, Patricia del Valle Tabernero1, Ana Díaz Rojo1, Andrea González Pigorini1, Esther Gigante Miravalles1, María Cristina Morante Perea1, Fernando Sabatel Pérez2 y Luis Rodríguez Padial1
1Cardiología. Hospital Universitario de Toledo, Toledo, España y 2Cardiología. Hospital Clínico San Cecilio, Granada, España.
- 5002-11. Caracterización y pronóstico de la amiloidosis cardiaca hereditaria por transtirretina en España
- Tomás Ripoll Vera1, Cristina Pericet Rodríguez2, José González Costello3, Ana José Manovel Sánchez4, Esther Zorio Grima5, Jaume Pons Llinares6, José Manuel García Pinilla7, Lucas Tojal Sierra8, Javier Limeres Freire9, Juan Ramón Gimeno Blanes10, Ana García Álvarez11, María Gallego Delgado12, José López Aguilera13, Rosa Macías Ruíz14 y M. Ángeles Espinosa Castro15
1Servicio de Cardiología. Hospital Universitario Son Llàtzer, Palma de Mallorca (Illes Balears), España, 2Servicio de Cardiología. Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), España, 3Servicio de Cardiología. Hospital Universitario de Bellvitge, Barcelona, España, 4Servicio de Cardiología. Hospital Juan Ramón Jiménez, Huelva, España, 5Servicio de Cardiología. Hospital Universitario La Fe, Valencia, España, 6Servicio de Cardiología. Hospital Son Espases, Palma de Mallorca (Illes Balears), España, 7Servicio de Cardiología. Hospital Clínico Universitario Virgen de la Victoria, Málaga, España, 8Servicio de Cardiología. Hospital Universitario Araba-Txagorritxu, Vitoria-Gasteiz (álava), España, 9Servicio de Cardiología. Hospital Universitario Vall d'Hebron, Barcelona, España, 10Servicio de Cardiología. Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, España, 11Servicio de Cardiología. Hospital Clínic, Barcelona, España, 12Servicio de Cardiología. Complejo Asistencial Universitario de Salamanca, Salamanca, España, 13Servicio de Cardiología. Hospital Universitario Reina Sofía, Córdoba, España, 14Servicio de Cardiología. Hospital Universitario Virgen de las Nieves, Granada, España y 15Servicio de Cardiología. Hospital General Universitario Gregorio Marañón, Madrid, España.
- 5002-12. Validación externa de la puntuación de riesgo de Columbia para predecir el pronóstico de los pacientes con amiloidosis cardiaca por transtirretina. Análisis del registro gallego de amiloidosis cardiaca
- Fausto de Andrés Cardelle1, Eduardo Barge Caballero1, Gonzalo Barge Caballero1, Eva González Babarro2, Andrea López López3, Inés Gómez Otero4, Raquel Bilbao Quesada5, Manuel López Pérez6, Mario Gutiérrez Feijoo7 y María G. Crespo Leiro1
1Complexo Hospitalario Universitario de A Coruña, A Coruña, España, 2Complejo Hospitalario de Pontevedra, Pontevedra, España, 3Hospital Universitario Lucus Augusti, Lugo, España, 4Complexo Hospitalario Universitario de Santiago de Compostela, Santiago de Compostela (A Coruña), España, 5Hospital Álvaro Cunqueiro, Vigo (Pontevedra), España, 6Hospital Arquitecto Marcide, Ferrol (A Coruña), España y 7Complexo Hospitalario, Ourense, España.
Más comunicaciones de los autores
- Artigas Baleri, Alicia
-
Badia Molins, Clara
- 4001-6 - Evaluación automática de mapas de deformación aórtica mediante tomografía computarizada 4D
- 4008-5 - Diferencias de género en el pronóstico aórtico en pacientes con síndrome de Marfan. Estudio longitudinal retrospectivo del registro REPAG
- 5002-6 - El strain de la aurícula izquierda predice eventos cardiovasculares adversos en la miocardiopatía hipertrófica: resultados preliminares de un estudio de RMC
- 6002-2 - Evaluación de la calidad de vida en el síndrome de Marfan
- 5002-8 - Evaluación de la capacidad funcional y parámetros cardiovasculares mediante CPET en pacientes con síndrome de Marfan
- 5002-3 - Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
- Belmar Clivillé, David
-
Campreciós Crespo, Marta
- 6019-100 - ¿Cómo se trata en Cataluña la obstrucción del tracto de salida del ventrículo izquierdo en la miocardiopatía hipertrófica?
- 5002-3 - Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
- 5021-6 - Remodelado ventricular tras implante de DAI en miocardiopatía dilatada: prevalencia, perfil clínico e impacto pronóstico
-
de Antonio Ferrer, Marta
- 5021-6 - Remodelado ventricular tras implante de DAI en miocardiopatía dilatada: prevalencia, perfil clínico e impacto pronóstico
- 6021-124 - Retirada farmacológica en pacientes con insuficiencia cardiaca avanzada: priorizando la calidad de vida
- 6122-10 - Infección del driveline y ducha en pacientes con dispositivos de asistencia ventricular izquierda de larga duración, mito o realidad
- 5002-3 - Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
- de Frutos Seminario, Fernando
-
Gayán Ordás, Jara
- 6109-9 - Caracterización del síndrome coronario agudo en función del nivel de lipoproteína (a)
- 5002-3 - Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
- 6019-100 - ¿Cómo se trata en Cataluña la obstrucción del tracto de salida del ventrículo izquierdo en la miocardiopatía hipertrófica?
- 4008-7 - Cohorte nacional de sospecha de miocarditis Pre-MYO: manejo asistencial en los 100 primeros casos
- 4014-6 - Utilidad del CA125 y NT-proBNP para evaluar la congestión en insuficiencia cardiaca crónica; subestudio del registro nacional CARDIOREN
-
Limeres Freire, Javier
- 5002-6 - El strain de la aurícula izquierda predice eventos cardiovasculares adversos en la miocardiopatía hipertrófica: resultados preliminares de un estudio de RMC
- 5002-11 - Caracterización y pronóstico de la amiloidosis cardiaca hereditaria por transtirretina en España
- 5002-3 - Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
- 5002-7 - Genotipos de alto riesgo en miocardiopatía dilatada: más allá de las arritmias
- 4008-5 - Diferencias de género en el pronóstico aórtico en pacientes con síndrome de Marfan. Estudio longitudinal retrospectivo del registro REPAG
-
Mirabet Pérez, Sonia
- 6069-435 - Empleo de estatinas en la vida real en receptores de trasplante cardiaco. Resultados del estudio multicéntrico DONOR-CAD
- 5021-6 - Remodelado ventricular tras implante de DAI en miocardiopatía dilatada: prevalencia, perfil clínico e impacto pronóstico
- 6104-2 - Beneficio de la colchicina en insuficiencia cardiaca aguda: diseño y características basales de un ensayo clínico aleatorizado, doble ciego, controlado con placebo (COLICA)
- 6122-10 - Infección del driveline y ducha en pacientes con dispositivos de asistencia ventricular izquierda de larga duración, mito o realidad
- 6021-124 - Retirada farmacológica en pacientes con insuficiencia cardiaca avanzada: priorizando la calidad de vida
- 5002-3 - Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
-
Moliner Abós, Carlos
- 5002-3 - Fenotipo y pronóstico de la miocardiopatía hipertrófica causada por p.(Gln892Lys) en MYH7: una variante endémica en Cataluña
- 5021-6 - Remodelado ventricular tras implante de DAI en miocardiopatía dilatada: prevalencia, perfil clínico e impacto pronóstico
- 6021-124 - Retirada farmacológica en pacientes con insuficiencia cardiaca avanzada: priorizando la calidad de vida
- Muñoz Cabello, Patricia
- Rodríguez Santiago, Benjamín
- Serra Juhè, Clara