El síndrome de Noonan (SN, OMIM 163950) es un trastorno genético multisistémico con una incidencia estimada relativamente alta, de alrededor de 1/1.000 a 1/2.500 nacidos vivos1.
Dicho síndrome constituye la causa sindrómica más frecuente de cardiopatía congénita después del síndrome de Down1. El diagnóstico del SN se basa principalmente en la identificación de unas manifestaciones clínicas características, como unos rasgos faciales distintivos, talla baja y defectos cardiacos congénitos1. Se producen anomalías cardiovasculares en un 50-90% de los individuos con SN, y la más frecuente es la estenosis de la válvula pulmonar. La miocardiopatía hipertrófica (MCH), que se observa en un 20-30% de los individuos, suele aparecer de manera temprana. Otras anomalías cardiovasculares descritas en el SN son la comunicación interauricular e interventricular, la coartación de la aorta, el canal auriculoventricular parcial y la tetralogía de Fallot1.
El SN se ha considerado clásicamente un trastorno autosómico dominante; sin embargo, muy recientemente se ha descrito un patrón de transmisión hereditaria autosómica recesiva asociada con variantes bialélicas en el regulador transcripcional 1 de tipo leucina-cremallera (zipper) 1 (LZTR1)2. Se presentan aquí 2 nuevos casos de SN de herencia autosómica recesiva.
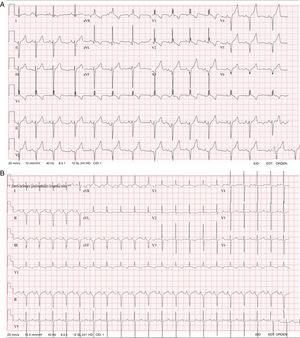
Caso 1. Paciente varón al que se diagnosticó al nacer una MCH grave y estenosis leve de la válvula pulmonar. Tenía rasgos faciales característicos de SN, con frente ancha, hipertelorismo, fisuras palpebrales inclinadas hacia abajo, orejas con rotación posterior y hélice engrosada, y un tórax ancho con cuello alado. En el electrocardiograma (ECG) se observaron complejos QRS anchos para su edad (> 0,10 s a la edad de 4 años), bloqueo de rama derecha, desviación del eje a la izquierda y un patrón negativo en las derivaciones precordiales izquierdas (figura 1A). Se solicitó un estudio genético para el análisis de RASopatías mediante secuenciación de nueva generación (NGS, panel de 18 genes), en las que se identificaron 2 nuevas variantes en el gen LTRZ1 (p.Arg362* y c.1149+1G>T).

Caso 2. Paciente varón al que se diagnosticó al nacer una MCH grave sin obstrucción. El ECG fue característico del SN, con complejos QRS anchos para la edad, desviación del eje a la izquierda y un patrón negativo en las derivaciones precordiales izquierdas (figura 1B). Presentó problemas graves de alimentación, que requirieron tratamiento mediante sonda nasogástrica y gastrostomía. Se realizó estudio genético con el mismo panel de RASopatías y se identificaron 2 nuevas variantes en el gen LTRZ1 (p.Val579Met y c.2070-2A>G).
La determinación del genotipo de los padres sanos, no consanguíneos, demostró la segregación genética en ambos casos clínicos, confirmando el estado bialélico de las variantes.
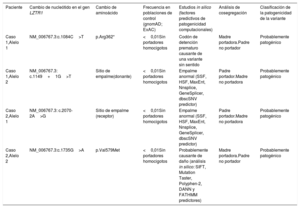
Los análisis clínicos y genéticos permitieron clasificar estas 4 variantes halladas en el gen LZTR1 como probablemente patogénicas (tabla 1). Una de las variantes identificada en el caso 1, la c.1149+1G>T, no se ha descrito anteriormente en la literatura, aunque sí se ha descrito otra variante que afectaba al mismo sitio de empalme (splicing) en heterocigosis compuesta en un paciente con una forma autosómica recesiva del SN2. La otra variante identificada en el caso 1, la c.1084C>T (p.Arg362*), introduce una señal de detención que conduce a un transcripto aberrante y, por consiguiente, no se traduciría. Por lo que respecta al caso 2, la variante c.2070-2A>G afecta a un sitio canónico de empalme en esta región intrónica del gen. Por último, la otra variante identificada en el caso 2, p.Val579Met, se encuentra en el dominio BACK I, en donde se han identificado otras variantes missense clasificadas como patógenicas. Serán necesarios estudios funcionales específicos para confirmar de manera definitiva su patogenicidad.
Clasificación de las variantes identificadas en el gen LZTR1
| Paciente | Cambio de nucleótido en el gen LZTR1 | Cambio de aminoácido | Frecuencia en poblaciones de control (gnomAD; ExAC) | Estudios in silico (factores predictivos de patogenicidad computacionales) | Análisis de cosegregación | Clasificación de la patogenicidad de la variante |
|---|---|---|---|---|---|---|
| Caso 1,Alelo 1 | NM_006767.3:c.1084C>T | p.Arg362* | <0,01Sin portadores homocigotos | Codón de detención prematuro causante de una variante sin sentido | Madre portadora.Padre no portador | Probablemente patogénico |
| Caso 1,Alelo 2 | NM_006767.3: c.1149+1G>T | Sitio de empalme(donante) | <0,01Sin portadores homocigotos | Empalme anormal (SSF, HSF, MaxEnt, Nnsplice, GeneSplicer, dbscSNV predictor) | Padre portador.Madre no portadora | Probablemente patogénico |
| Caso 2,Alelo 1 | NM_006767.3: c.2070-2A>G | Sitio de empalme (receptor) | <0,01Sin portadores homocigotos | Empalme anormal (SSF, HSF, MaxEnt, Nnsplice, GeneSplicer, dbscSNV predictor) | Padre portador.Madre no portadora | Probablemente patogénico |
| Caso 2,Alelo 2 | NM_006767.3:c.1735G>A | p.Val579Met | <0,01Sin portadores homocigotos | Probablemente causante de daño (análisis in silico: SIFT, Mutation Taster, Polyphen-2, DANN y FATHMM predictores) | Madre portadora.Padre no portador | Probablemente patogénico |
Estos 2 casos clínicos que se presentan resultan de especial interés tanto para el diagnóstico clínico como para el asesoramiento genético.
En primer lugar, se trata de 2 nuevos casos clínicos de una forma autosómica recesiva del SN. Este patrón de herencia fue propuesto hace 5 décadas por Dieckman et al.3, quienes describieron a 2 hermanos y 1 hermana con manifestaciones clínicas de SN consistentes en MCH y pterygium colli, sin que ninguno de los dos progenitores estuviera afectado. Sin embargo, no ha sido hasta hace muy poco cuando los datos clínicos y genéticos han confirmado la existencia de una forma de SN que se hereda siguiendo un patrón autosómico recesivo, cuando Johnston et al.2 describieron unas variantes patógenicas bialélicas en el gen LZTR1 en 23 niños con diagnóstico clínico de SN y padres heterocigotos sin afectación. Anteriormente se habían descrito variantes en línea germinal en el gen LTZR1 asociadas con un SN autosómico dominante con una expresividad clínica muy variable4. Estos datos indican que las variantes patogénicas en línea germinal de este gen podrían ser causa tanto de un SN dominante como de uno recesivo.
Otro aspecto que cabe resaltar en estos 2 casos que se presentan son las características observadas en el ECG que se habían descrito hace décadas como típicas del SN, caracterizados por complejos QRS anchos, con un patrón predominantemente negativo en las derivaciones precordiales izquierdas, desviación del eje a la izquierda y ondas Q5. Otros autores han observado este patrón de ECG en casi el 60% de los pacientes con SN, con independencia de la presencia o ausencia de defectos anatómicos cardiacos6. Esto indica que el ECG continúa siendo una herramienta muy útil para respaldar una sospecha diagnóstica de síndrome de Noonan y debe realizarse en todos los pacientes con un fenotipo indicativo de SN.
Es importante buscar cualquier indicio que pueda corroborar la sospecha de este síndrome, ya que el diagnóstico de SN continúa siendo clínico basado en la observación de unas manifestaciones relevantes, teniendo en cuenta que en hasta un 25% de los pacientes evaluados no se encuentra una mutación causal genética. De todos modos, cabe prever que, en un futuro próximo, en muchos de estos casos se obtenga una confirmación genética al ampliar las pruebas moleculares a estas variantes patogénicas recientemente descritas.
CONFLICTO DE INTERESESL. Monserrat es accionista de Health in Code S.L. J.P. Trujillo-Quintero trabaja en Health in Code S.L. Los demás autores han indicado que no tienen posibles conflictos de intereses que declarar.