
Las complicaciones cardiovasculares tienen una alta prevalencia en los pacientes con COVID-19 y son motivo frecuente de hospitalización, mortalidad y secuelas. En está revisión se describen los principales mecanismos fisiopatológicos implicados en la aparición de estas complicaciones. Tras la viremia inicial, se produce una infiltración y reproducción en los pulmónes, con activación del sistema inmunitario, liberación de citocinas y generación de un estado proinflamatorio con sepsis y fallo multiorgánico. El daño miocárdico puede deberse a una afección viral directa con respuesta inflamatoria local, o indirectamente a una inflamación sistémica inapropiada con marcada liberación de citocinas. Además, se genera un estado protrombótico que, junto con la afección viral vascular, pueden desencadenar eventos trombóticos e isquémicos secundarios a daño microvascular o inestabilización de placas de ateroma previas. Son necesarios nuevos estudios para esclarecer la fisiopatología tras estos eventos cardiovasculares y contribuir al desarrollo de nuevos tratamientos efectivos.
Palabras clave
Las complicaciones cardiovasculares tienen gran prevalencia en los pacientes con COVID-19 (el 30% de los hospitalizados)1, son motivo frecuente de hospitalizaciónes, reingresos y morbimortalidad y pueden originar secuelas que requerirán seguimiento a largo plazo1-5.
Coronavirus del síndrome respiratorio agudo grave de tipo 2 (SARS-CoV-2)Los coronavirus son un grupo amplio de virus envueltos de material genético como el ácido ribonucleico (ARN), de entre 26 y 32 kb de longitud, dentro de la familia Coronaviridae. Hay 4 géneros en la sub-familia Orthocoronavirinae, los alfacoronavirus, betacoronavirus, gammacoronavirus y deltacoronavirus. De estos, los alfacoronavirus y betacoronavirus infectan a los mamiferos, mientras que los gammacoronavirus y deltacoronavirus infectan a las aves6. Hay 7 coronavirus que infectan a los seres humanos: los alfacoronavirus HCoV-NL63 y 229E, que tienden a causar una enfermedad leve en los adultos; el virus del síndrome respiratorio del Oriente Medio (MERS-CoV) y el virus del síndrome respiratorio agudo grave (SARS) de los betacoronavirus, que causan una enfermedad respiratoria grave, y el OC43 y el HKU1, que están asociados con una enfermedad leve. La COVID-19 tiene origen en un nuevo betacoronavirus7, denominado coronavirus 2 del síndrome respiratorio agudo grave (SARS-CoV-2). El SARS-CoV-2 tiene un genoma que coincide en un 96% con el de un coronavirus de murciélago similar al SARS, lo que indica un origen zoonotico de la infección8. Una descripcion de los principales tipos de coronavirus que infectan a la especie humana se expone en la figura 19.

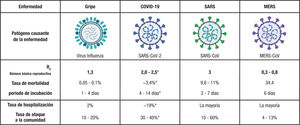
Comparacion epidemiologica de las infecciónes respiratorias virales, con datos disponibles en marzo de 2020. Modificada con permiso de BioRender9. R0 o número reproductive básico es un parametro teorico que proporciona cierta información acerca de la velocidad con que una enfermedad puede propagarse en una población determinada. MERS-CoV: coronavirus del síndrome respiratorio del Oriente Medio; SARS-CoV: coronavirus del síndrome respiratorio agudo grave.
La transmision de está enfermedad se basa principalmente en la exposicion de una persona sana a objetos contaminados o a personas infectadas, que incluso pueden encontrarse asintomaticas10. El periodo de incubación del SARS-CoV-2 es de 5 días, pero puede exten-derse hasta 14 días11.
El virus entra al organismo a través de las mucosas (oral, nasal o conjuntiva). La proteína viral S es capaz de mediar la unión con el receptor y fusionarse con la membrana celular de las células epiteliales del pulmón. Análisis comparativos entre las proteínas S del SARS-CoV y el SARS-CoV-2 muestran que el genoma de estas proteínas coincide en un 80%, lo que indica que utilizan el mismo receptor para entrar en la célula y poder reproducirse7: la enzima de conversión de la angiotensina 2 (ECA-2)12. Una vez que ocurre la unión entre la superficie del virus y la membrana celular de la célula huésped, comienza un proceso de fusión entre la membrana vírica y la plasmática. Posteriormente, el ARN del virus comienza a transcribirse y reproducirse, procesos que ocurren principalmente dentro de las células epiteliales del tracto respiratorio superior e inferior13. SARS-CoV-2 causa un daño directo en los epitelios pulmonares que puede conducir a una neumonía grave y al síndrome de dificultad res-piratoria del adulto (SDRA).
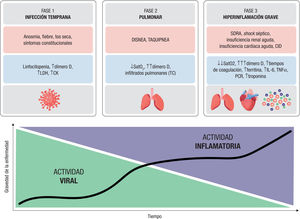
La progresión de la enfermedad a lo largo del tiempo se divide en 3 fases patológicas (figura 2): la fase de infección temprana, una fase pulmonar y una fase de hiperinflamación grave. La fase de infección temprana se caracteriza por la infiltración y duplicación viral. La linfocitopenia es un hallazgo de laboratorio clave en está fase. La enfermedad progresa hasta la fase pulmonar, caracterizada por afección respi-ratoria y alteración de las pruebas de imagen torácica. La última fase de hiperinflamación se caracteriza por una respuesta inflamatoria exagerada, impulsada por la inmunidad del huésped, que puede conducir a fallo multiorgánico y coagulación intravascular diseminada (CID) en ciertos pacientes14. Además, la hipoxia observada en pacien-tes con neumonía grave y SDRA también pueden conducir a un mayor daño secundario de los órganos y la muerte de los pacientes en estado crítico.

Fases de la COVID-19. Se muestran las 3 fases de la infección con sus síntomas y los marcadores de laboratorio y de imagen característicos de cada etapa. En la gráfica se pone de manifiesto la relación entre estas fases y la actividad viral e inflamatoria. Figura de elaboracion propia. CID: ; CK: creatincinasa; IL: interleucina; LDH. lactato deshidrogenasa; PCR: proteina C reactiva; SatO2: síndrome arterial de oxigeno; SDRA: síndrome de dificultad respiratoria del adulto; T’: tiempos; TC: tomografía computarizada; TNF: factor de necrosis tumoral.
A continuación se describen los distintos mecanismos fisiopatológicos implicados en el desarrollo de complicaciones cardiovasculares de la COVID-19. Se trata de hipótesis basadas en los datos clínicos y la evidencia previa acerca de la respuesta del sistema inmunitario a las infecciónes por otros coronavirus. No se trata de mecanismos exclu-yentes, y puede que sus sinergias e interacciones sean la causa de las complicaciones observadas (figura 3).

El daño miocárdico en los pacientes con COVID-19 tiene una elevada prevalencia y se ha relacionado con la gravedad de está enfermedad1,2,4. Entre un 7 y un 28% de los pacientes tienen elevación de troponina34,15-17 y valores aumentados al ingreso que se mantienen durante la hospitalización se han asociado con riesgo de eventos adversos hospitalarios (necesidad de ventilación mecánica, aparición de arritmias y muerte)18. También se ha objetivado una importante proporción de pacientes con péptidos natriuréticos aumentados, y la combinacion de ambos marcadores es pronostica de necesidad de ingreso en unidad de cuidados intensivos, ventilación mecánica y muerte19. Se ha documentado daño miocárdico en cualquiera de las 3 fases de la enfermedad y su fisiopatología es diversa e incierta. Los mecanismos por los que el SARS-CoV-2 podría ocasionar daño mio-cárdico son:
Daño miocárdico directo por la infección viralLa miocarditis aguda es una complicación de las infecciónes virales bien conocida. La comunicación de diversos casos indica que la miocarditis fulminante es uno de los potenciales eventos adversos de la COVID-195, 20. Sin embargo, este mecanismo no está claramente establecido y podría ser multifactorial.
El daño miocárdico directo del SARS-CoV-2 en los cardiomiocitos no es un mecanismo claramente demostrado. En China las autopsias no han identificado particulas virales en las biopsias miocárdicas21, mientras que estudios previos de autopsias de pacientes fallecidos por SARS en 2002 sí objetivaron ARN viral hasta en un 35% de las muestras22. Este mecanismo podría ser compartido por el SARS-CoV-2, pues ambos virus tienen un genoma parecido18,23. sí se ha visto necrosis de cardiomiocitos e infiltrado mononuclear en varias autopsias de pacientes fallecidos por la COVID-1921. La infección por el SARS-CoV-2 podría ser directa de cardiomiocitos mediada por los receptores de la ECA-2, con lisis celular y activation de la respuesta inmunitaria innata con liberación de citocinas proinflamatorias. Las proteínas liberadas por la lisis celular mostrarían epítopos similares a los antígenos virales y activarían la inmunidad adquirida mediada por anticuerpos y linfocitos T. Los linfocitos, a su vez, estimularían la cascada inflamatoria y la citolisis. Además, se produciria una migración de macrófagos, causa de la inflamación crónica con disfunción ventricular24. Lainvasión del virus a través de los receptores de la ECA-2 no daria al virus solo la entrada en la célula, sino también una disminución de la expresión de estos receptores con disminución de la conversión de la angiotensina II en angiotensina 1-7 y disminución de los efectos protectores cardiovasculares derivados25.
Daño miocárdico secundario a hipoxemia por insuficiencia respiratoriaEn la etapa temprana de la enfermedad, el virus infiltra el parénquima pulmonar y comienza a proliferar. En esos momentos la enfermedad cursa con síntomas constitucionales leves, dados por la activación de la inmunidad innata, fundamentalmente monocitos y macrófagos. Esto lleva a un daño tisular y procesos inflamatorios secundarios con vasodilatacion, permeabilidad endotelial y reclutamiento leucocitario, todo ello seguido de mayor daño pulmonar, hipoxemia y estrés cardiovascular26, 27, que pueden ser causa de elevación de marcadores de daño miocárdico como traducción de un daño miocárdico subyacente. En un subconjunto de pacientes, está respuesta inmunitaria continúa amplificándose, lo que resulta en una inflamación sistémica26,27.
Respuesta inmunitaria e inflamación secundaria a infección (tormenta de citocinas)Lainvasión celular por el SARS-CoV-2 provoca una reacción inmunitaria ineficaz pero amplificada, con una liberación de citocinas inflamatorias que puede resultar en reacción inflamatoria sistémica, sepsis y daño multiorgánico27. La existencia de miocarditis en la COVID-19 sin infiltración viral puede ser la manifestacion de la afeccion cardiaca por está inflamación sistémica28.
Tras los pulmones, los órganos inmunológicos son el segundo sistema más afectado. En los ganglios linfáticos, se produce una disminución de linfocitos CD4+ y CD8+, y se objetiva linfocitopenia en sangre periférica21. Es especialmente llamativa la disminución de las células T reguladoras, que tienen un papel crítico en la homeostasis del sistema inmunitario y la prevencion de una excesiva inflamación tras la infección26’29,30. Además, se produce una activación inefectiva de los linfocitos T citotóxicos CD8+ y los linfocitos T natural killer, con una aclaramiento viral inefectivo y producción débil de anticuerpos. La reducción de linfocitos T CD4+ y CD8+ produce una activación de macrófagos con una relativa dominancia de células mononuclerares (monocitos y macrófagos) en los tejidos dañados y una respuesta inmunitaria descontrolada e ineficaz, con un síndrome de liberación de citocinas31.
En los pacientes con la COVID-19, se ha visto aumento de interleucina (IL) 1ß, IL-6, interferon gamma (IFNγ), proteína 10 inducible por IFNγ, proteína 1 de atraccion de monocitos, factor estimulador de colonias de granulocitos, proteína 1a inflamatoria de macrófagos y factor de necrosis tumoral alfa, entre otros32,33. Estas citocinas activan senales que perpetuan la inflamación y se relacionan con la gravedad de la enfermedad32,33.
La IL-6 hace un papel fundamental, y es un importante predictor de mortalidad34. Además, es un biomarcador que se ha relacionado con morbimortalidad cardiovascular en relación con ateroesclerosis35. La tormenta de citocinas con aumento de IL-6 que se observa en algu-nos pacientes podría tener consecuencias cardiovasculares importantes al causar taquicardia, hipotensión y disfunción ventricular. También se ha visto cardiotoxicidad secundaria que se manifiesta como eventos arrítmicos y elevación de marcadores de daño miocárdico y podría estar implicada en eventos a largo plazo, como fenómenos ate-roescleroticos36,37, fibrosis cardiaca38, remodelado vascular con hiper-tension pulmonar39 y riesgo cardiovascular aumentado40.
Estado protrombóticoLas complicaciones trombóticas están emergiendo como secuelas importantes que contribuyen a morbimortalidad significativa41. sí bien la infección por SARS-CoV-2 suele producir un cuadro seudogri-pal leve, en un bajo porcentaje se presentara como una neumonía que puede combinarse con un estado de CID en los casos más graves32. Se han documentado criterios diagnósticos de CID hasta en un 71% de los pacientes fallecidos42, lo que se explicaría por el estado crítico de estos pacientes. Sin embargo, datos analíticos indicadores de CID, como cifras elevadas de dímero D y productos de degradacion de fibrina, son muy prevalentes y pueden observarse desde estadios tempranos de la enfermedad43. Por otro lado, no solo las formas graves de CID se han relacionado con la COVID-19, sino también otros fenómenos trombóticos como la embolia pulmonar, la trombosis venosa profunda, el accidente cerebrovascular isquémico y el infarto agudo de miocardio (IAM)41, 43-45.
Se han publicado varios estudios que informan de que el comportamiento de las variables de coagulación y el recuento de plaquetas en estos pacientes indica un estado protrombótico de hipercoagula-cion43. Se desconoce sí estos cambios son un efecto específico del SARS-CoV-2 o son una consecuencia de la tormenta de citocinas asociada con el síndrome de respuesta inflamatoria sistémica46. A continuación se detallan algunos de estos cambios.
Cifras de dímero D aumentadas. La proporción de pacientes con dímero D aumentado oscila entre el 14 y el 46%43. Metanálisis recientes43,47,48 coinciden en una relación directa entre el dímero D y la gravedad de la COVID19 considerada como la necesidad de ventilación mecánica, ingreso en unidad de cuidados intensivos o muerte. Además, estas alteraciones se presentan antes de la etapa de progresión rápida, lo cual podría ser un marcador incipiente de enfermedad grave6.
Trombocitopenia leve. Se ha detectado en un 5-18% de estos pacientes, normalmente leve (media, 100.000 plaquetas/pl)43 y relacionada con la gravedad43,47,48.
Prolongación del tiempo de protrombina. Solo un 2-11% de la población estudiada presenta prolongación del tiempo de protrombina durante la hospitalización. Se asocia con la gravedad de la enfermedad43,48.
Tiempo de tromboplastina parcial activado. Se trata de una alteración controvertida. Existen variaciones en los tiempos de tromboplastina parcial activados en un 6-26% de estos pacientes, pero pueden tanto aumentar como reducirse43. No está clara su relación con la gravedad de la enfermedad43.
Los mecanismos fisiopatológicos de la coagulopatía parecen atender a múltiples vías interrelacionadas entre sí con complejos mecanismos en los que intervienen tanto elementos celulares como plasmáticos de los sistemas hemostático e inmunitario. A continuación, se describen los principales:
Regulación negativa de la expresión de ECA2La ECA2 participa en el sistema renina-angiotensina-aldosterona catalizando la conversión de la angiotensina II a angiotensina 1-7. Esta, mediante su unión a los receptores AT1, se opone a las acciones vasoconstrictoras, proinflamatorias, prooxidantes, proproliferativas y profibróticas ejercidas por la angiotensina II49. La regulación negativa de la expresión de ECA2 produce un estado proinflamatorio y prooxidativo que contribuye de manera directa al estado protrombótico50.
Daño directo por infección viral a nivel vascularEl músculo liso vascular y el endotelio presentan en su membrana el receptor de ECA-2, por lo que es un tejido con invasión y proliferación viral demostrada51. La proliferación viral y el daño celular a este nivel produciría la activación de macrófagos que liberan citocinas (principalmente IL-1 ß e IL-6), que promueve la expresión de moléculas de adhesión para la activación endotelial, la infiltración de células inflamatorias y la inflamación vascular que da lugar a una vasculitis aguda51. Además, las células musculares lisas y el endotelio también liberarían factores procoagulantes como el plasminógeno y citocinas proinflamatorias que contribuyen a la propagación de las lesiones microcirculatorias52. Por otro lado, se da una gran expresión de ECA2 en el pericito, lo cual se relaciona directamente con un estado de disfunción endotelial que conlleva la situación de hiperactividad plaquetaria y procoagulante que dará lugar a microangiopatía y microtrombos en diferentes órganos, lo que exacerba aun más el estado de disfunción multiorgánica6,51. La vasculitis no es casual, y ya se ha relacionado la enfermedad de Kawasaki con otras infecciónes por coronavirus53. Además, el SARS ha documentado vasculitis sistémica54, y en pacientes con COVID-19 se han documentado lesiones vasculíticas digitales y se ha considerado la vasculitis con daño microvascular como una causa importante del daño miocárdico y renal49.
Inflamacion sistémica y tormenta de citocinasEs uno de los mecanismos más aceptados para explicar la asociación de la COVID-19 con los eventos trombóticos y está presente en el 80-100% de los casos complicados de COVID-1943. Diversos trabajos de investigación en el seno de neumonías graves con inflamación sistémica objetivan alteraciones en los 3 niveles de la hemostasia (sistema de coagulacion, actividad plaquetaria y función vascular)43. En el seno de una neumonía (incluso sin sepsis) se han observado anomalías en la coagulación sistémica, incluida la activación de la coagulación y la inhibición de los factores anticoagulantes (proteína C), con normalización de ellos tras la resolución del cuadro respiratorio55. También se ha objetivado aumento de la activación plaquetaria, reflejado analíticamente como elevación de CD40, P-selectina y tromboxano plaquetario, que da lugar a un estado de hiperactividad plaquetaria que favorece la coagulopatía. Finalmente, en el sistema vascular, se ha relacionado la neumonía con alteraciones transitorias del tono vasomotor arterial por la inactivación del óxido nítrico y la producción de eicosanoides plaquetarios56. Como ya se ha mencionado, la infección viral induce una reacción inmunitaria excesiva en el huésped y una «tormenta de citocinas» con incremento de IL-6, entre otras. está ejerce funciones nocivas, como la hiperpermeabilidad capilar causante del edema intersticial57, la inflamación pulmonar causante de la fibrinolisis pulmonar que aumenta el dímero D y la activación endotelial, plaquetaria y linfocitaria que lleva al desequilibrio en la producción de trombina, con depósito de fibrina que origina la microangiopatía y el daño tisular58. La relación directa entre la reacción inmunitaria excesiva y el estado protrombótico se pone de manifiesto por la asociación de las concentraciones de proteína C reactiva con el dímero D y los eventos trombóticos59.
Estrés oxidativoLas neumonías, principalmente en situaciones de inflamación sistémica, hiperactiva las cascadas de producción de radicales libres de oxígeno a través de la vía de la nicotinamida adenina dinucleótido fosfato (NADPH) oxidasa 2 (NOX2). Estos radicales libres de oxígeno están implicados tanto en la coagulación como en la activación plaquetaria y actúan como señal para promover la generación de trombina o la agregación plaquetaria o inhibir la dilatación arterial43. Todavía faltan datos sobre la actividad de la NOX2 en pacientes con SARS-CoV-2 pero, por extrapolación del conocimiento de la regulación positiva de NOX2 en la inflamación sistémica y la patogenicidad de otros virus de ARN60, parece plausible que este mecanismo pueda ser una de las vías fisiopatológicas de los eventos trombóticos asocia-dos con la COVID-19.
Otros mecanismosLos pacientes con COVID-19 inmovilizados presentan una situa-cion de estasis venosa que, junto con el estado protrombótico y la dis-funcion endotelial ya detallada, cumplen los 3 criterios de la tríada de Virchow y supone un alto riesgo de enfermedad tromboembolica venosa. Por otro lado, los tratamientos farmacológicos empleados pueden tener interacciones farmacológicas adversas que reduzcan la efectividad de los antiagregantes plaquetarios y los anticoagulantes y aumenten el riesgo de eventos trombóticos. Ese es el caso del lopinavir/ritonavir, que como inhibidor del citocromo CYP3A4 puede reducir la efectividad del clopidogrel y como inductor del CYP2C9 puede reducir la concentración plasmática de los antagonistas de la vitamina K41. Por último, el estado de pandemia genera miedo y desinformación que pueden llevar a la falsa percepción de que los fármacos antitrombóticos confieren un mayor riesgo de padecer la COVID-19, y que algunos pacientes interrumpan la anticoagulación, con el riesgo que conlleva41.
Isquemia miocárdicaEntre las consecuencias del estado proinflamatorio y del estado protrombótico, pueden producirse eventos isquémicos como la isquemia miocárdica con daño miocárdico secundario. Este no solo se debe a un síndrome coronario agudo trombotico (IAM de tipo 1), sino que en muchas ocasiones es secundario a un desequilibrio entre la demanda y la oferta miocárdicas de oxígeno (IAM de tipo 2) en el contexto de está infección y su respuesta inmunitaria. Gracias a los estu-dios clínicos y en el seno de otras infecciónes virales, se sabe que los pacientes con enfermedad coronaria previa y aquellos con factores de riesgo de enfermedad cardiovascular ateroesclerótica están en mayor riesgo de sufrir una isquemia miocárdica durante las infecciónes agudas61,62. Los mecanismos fisiopatológicos subyacentes son diversos y complejos, con diferentes efectores y con vías interrelacionadas.
Desequilibrio entre oferta y demanda (IAM de tipo 2)Una de las principales causas de isquemia miocárdica en la COVID-19 es el desequilibrio entre el suministro y la demanda miocardicos de oxígeno. Por un lado, la reducción en la oferta de oxígeno al miocardio generalmente tiene origen en la insuficiencia respiratoria hipóxica, sí bien en situaciones graves la hipotensión arterial también puede ser relevante. En el aumento de la demanda, se han descrito diferentes mecanismos, como fiebre, taquicardia y estimulación simpatica, que producen aumentos de tension de la pared, contraccion o frecuencia cardiaca que dan lugar a un aumento de la demanda mio-cardica de oxígeno50,63. Conforme la enfermedad progresa, este desequilibrio se acentua cada vez más, lo que puede producir lesiones miocárdicas, sobre todo en pacientes con enfermedad coronaria subyacente que ya han agotado la capacidad de reserva miocárdica en el lado de la oferta63.
Rotura de placa ateroesclerótica (IAM de tipo 1)El IAM de tipo 1 es el causado por la rotura de una placa ateroes-clerotica de la que se forma un trombo intracoronario que ocluye al menos parcialmente la luz. está entidad también se ha relacionado con la COVID-1963. En este proceso están implicados la tormenta de citocinas y el estrés inflamatorio sistémico grave, que inducen un estado protrombótico que genera inestabilidad y rotura de la placa ateroesclerotica, así como la supresion de la expresión de la ECA2 por el aumento de de la angiotensina II, que origina el estrés oxidativo, la disfunción endotelial y la vasoconstriccion relacionadas con la inestabilidad de la placa. Por último, no se puede obviar el efecto del daño viral directo en las células endoteliales y pericitos vasculares, que también conduce a la inestabilidad de la placa1.
Afección microvascular coronariaComo ya se ha detallado, existe un daño vascular (vasculitis aguda) que, junto con el estado protrombótico y la hiperactividad plaqueta-ria, puede dar lugar a una microangiopatía y microtrombosis también en la microvasculatura coronaria6,51. En este contexto, se postula un papel principal del daño a los pericitos y las células endoteliales, ya sea por daño viral directo o indirecto a través de las citocinas y el estado inflamatorio sistémico. Se puede producir una alteración de la función endotelial con interrupcion de la microcirculacion coronaria y con consecuencias isquémicas, sí bien se trata de un mecanismo hipotético1.
ConclusionesLos mecanismos exactos implicados en la fisiopatología de las complicaciones cardiovasculares de los pacientes con COVID-19 aun están en estudio. La evidencia publicada, tanto derivada de la observación clínica de la infección por el SARS-CoV-2 como los datos previos de infecciónes por otros coronavirus, muestra que, ademas del daño miocárdico directo, hay una serie de mecanismos desencade-nados por la activación anormal y amplificada del sistema inmunitario que genera una tormenta de citocinas y un estado proinflamatorio que se asocia con un estado de hipercoagulabilidad, fenómenos protromboticos y eventos isquémicos secundarios. La gravedad de la neumonía con hipoxia grave e incluso hipotensión concomitante en los casos más graves también contribuyen al desarrollo de estas complicaciones. Se requiere, por lo tanto, un estudio más en profundidad para comprender la fisiopatología de estos eventos y contribuir al desarrollo de nuevos tratamientos efectivos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Información sobre el suplementoEste artículo forma parte del suplemento titulado «COVID-19 y enfermedad cardiovascular. Un nuevo reto para la cardiologia», que ha sido patrocinado por Boehringer Ingelheim España.