
La cardiopatía isquémica continúa siendo responsable de una gran morbilidad y mortalidad a nivel poblacional. Con el envejecimiento de la población se espera que el número de casos aumente en los próximos años y su prevención es un elemento importante en las políticas sanitarias. El riesgo de presentar cardiopatía isquémica está relacionado con características genéticas del individuo y su interacción con factores ambientales y estilos de vida. En los últimos años se ha avanzado considerablemente en el conocimiento de las bases genéticas de la cardiopatía isquémica. En este artículo se presenta una revisión narrativa sobre el conocimiento actual de la genética de esta patología y sus implicaciones clínicas: la identificación de dianas terapéuticas, el estudio de la relación causal entre un biomarcador y una enfermedad, la mejora en la predicción del riesgo y la identificación de pacientes respondedores o no respondedores a un tratamiento farmacológico.
Palabras clave
Las enfermedades cardiovasculares —en general— y la cardiopatía isquémica (CI) —en particular— continúan siendo las principales responsables de la mortalidad y la morbilidad mundial en 2015 y explican el 14,1 y el 6,7% respectivamente del total de años de vida ajustados por discapacidad1. La CI es una enfermedad compleja y multifactorial relacionada con factores ambientales que incluyen los estilos de vida (dieta, actividad física, tabaquismo), factores genéticos y la interacción entre ellos2.
En los últimos 10 años hemos asistido a avances significativos en el conocimiento de la arquitectura genética de la CI, y también a su traslación a la práctica clínica3. Los objetivos de esta revisión narrativa son resumir los conocimientos actuales sobre las bases genéticas de la CI; presentar algunos ejemplos de su impacto en la práctica clínica, y plantear algunos retos para el futuro.
LA ARQUITECTURA GENÉTICA DE LA CARDIOPATÍA ISQUÉMICACuando hablamos de la arquitectura genética de un fenotipo (una característica biológica cuantificable) nos referimos a qué genes y, dentro de estos genes, qué variantes genéticas determinan o se asocian con el fenotipo de interés.
Pero antes de iniciar el estudio de la arquitectura genética de un fenotipo hay que evaluar si el componente genético es importante o no. En el caso de la CI, varios estudios de cohortes han identificado que los antecedentes familiares de CI se asocian con un mayor riesgo de presentar la enfermedad4,5; lo que sugiere la importancia de los factores genéticos (aunque también hay que considerar que la familia no solo transmite información genética, sino también valores y estilos de vida). La heredabilidad de un fenotipo informa sobre el porcentaje de variabilidad de dicho fenotipo, que está relacionado con la variabilidad genética de la población6. Algunos estudios han cuantificado la heredabilidad de la CI, que oscilaría entre el 35 y el 55%7–9.
Una vez demostrada la relevancia del componente genético, se pueden diseñar estudios para identificar los genes y las variantes genéticas que se asocian con la CI. Desde el punto de vista de la trasmisión genética se puede considerar que hay 2 grandes grupos de fenotipos o enfermedades:
- •
Monogénicas (u oligogénicas), en las que el riesgo de presentar una enfermedad está relacionado con la presencia de variantes genéticas en uno o en un número reducido de genes. Un ejemplo claro es la hipercolesterolemia familiar en la que se han identificado un grupo de genes (LDLR, APOB, PCSK9 y LDLRAP1) que presenta variantes genéticas que determinan la presentación de esta alteración10,11.
- •
Poligénicas o complejas, en las que el riesgo está determinado por múltiples genes, muchas variantes genéticas en estos genes y su interacción con factores ambientales12. La CI es un ejemplo claro de este tipo de fenotipos complejos.
Para el estudio de la arquitectura genética de un fenotipo se pueden utilizar 4 aproximaciones diferentes:
- 1.
Estudios de ligamiento. Este tipo de estudios han demostrado ser útiles para el estudio de enfermedades mono-oligogénicas. Se realizan en familias en los que existe al menos 1 caso que presenta la enfermedad (índice) y familiares con y sin la enfermedad en más de una generación13,14. Se determinan unos centenares de marcadores genéticos repartidos por todo el genoma; se analiza su transmisión de una generación a otra, y si su presencia se asocia con la aparición de la enfermedad en estudio (segregación). El objetivo es identificar la región del genoma en la que se localiza el gen y la variante genética que causa la enfermedad. Una vez identificada dicha región se realizan más estudios en ella (genotipado o normalmente secuenciación) para identificar con precisión el gen y la variante causal. Este tipo de estudios ha sido muy útil en enfermedades mono-oligogénicas como la hipercolesterolemia familiar, en la que, por ejemplo, se identificó inicialmente una región en el cromosoma 1 en la que se encuentra el gen PCSK915 y posteriormente, mediante secuenciación, se identificaron en dicha región unas variantes genéticas en el gen PCSK9 que causaban esta enfermedad16. En el caso de enfermedades complejas, este tipo de estudios no ha sido tan útil17. No obstante, se han identificado variantes genéticas en los genes ALOX5AP18 y MEF2A19 asociadas con la CI.
- 2.
Estudios de asociación de gen candidato. En este tipo de estudio se utiliza clásicamente un diseño de casos y controles, y se analiza si la presencia de una o más variantes genéticas en un gen concreto es diferente en individuos con o sin la enfermedad. La selección de gen que se va a estudiar se basa en una hipótesis a priori que se fundamenta en el conocimiento fisiopatológico de la enfermedad y las variantes genéticas analizadas suelen ser variantes comunes (frecuencia alélica > 5%). Este tipo de estudios tampoco ha contribuido de forma significativa al conocimiento de la arquitectura genética de la CI ni tampoco de otros fenotipos complejos20. El principal problema de esta aproximación ha sido la falta de replicación de los resultados, que generalmente tiene relación con el pequeño tamaño de la muestra de los estudios que limitaba la potencia estadística para identificar variantes con magnitud de asociación pequeña.
- 3.
Estudios de asociación del genoma completo (GWAS [genome-wide association study]). A principios de este siglo, y con la aparición de nuevas tecnologías que permitían secuenciar el genoma o genotipar un gran número de variantes genéticas en la misma muestra, se produjo un gran avance que ha permitido progresar en el conocimiento de las bases genéticas de las enfermedades complejas. Además, se publicaron los resultados del estudio HapMap21,22 que observó que muchas variantes genéticas comunes están asociadas entre sí a nivel poblacional (están en desequilibrio de ligamiento). El conocimiento del patrón del desequilibrio de ligamiento del genoma humano, junto con el avance tecnológico, permitió diseñar kits de laboratorio que incluían entre 100.000 y 500.000 variantes genéticas y que capturaban gran parte de la variabilidad genética común del genoma humano y se diseñaron los estudios de GWAS. En este tipo de estudios se estudian centenares de miles de características genéticas y su relación con el fenotipo de interés. Se caracterizan por no tener hipótesis previa. La falta de esta hipótesis de partida tiene 2 grandes implicaciones a la hora de diseñar e interpretar los resultados de estos estudios: a) se exige que haya una muestra de descubrimiento, en la que se identifican una serie de variantes genéticas que potencialmente se asocian con el fenotipo de interés, y una muestra independiente de validación y replicación de los resultados observados en la muestra inicial, y b) al analizar de forma simultánea centenares de miles de variantes genéticas, se penaliza el estudio por el número de comparaciones múltiples realizadas y el valor de p que se considera estadísticamente significativo suele ser < 1·10–8.
Los estudios iniciales ya indicaron que la magnitud de la asociación entre las variantes genéticas comunes y los fenotipos complejos de interés son pequeñas, con odds ratio (OR) en el rango de 1,1 a 1,4. Para identificar variantes con esta magnitud de asociación, con un valor de p tan pequeño, y replicarlas en muestras independientes ha sido necesaria la colaboración internacional entre grupos de investigadores de todo el mundo para conseguir muestras de decenas de miles de individuos.
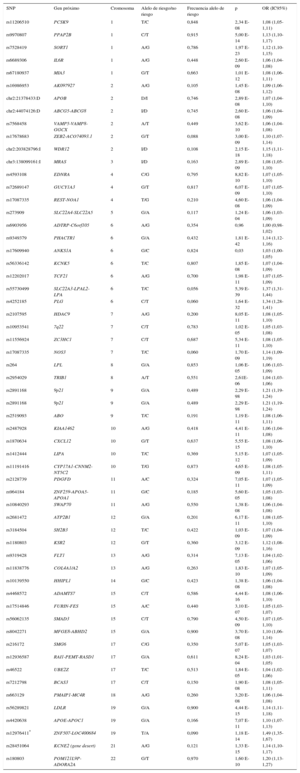
En el caso de la CI, los 2 primeros GWAS que se publicaron fueron consistentes e identificaron unas variantes en el cromosoma 9 que se asociaban con mayor riesgo de CI23,24. Posteriormente, se han publicado varios estudios GWAS25 y en 2015 se publicó un análisis conjunto de todos ellos que ha permitido identificar 55 loci, cada uno de ellos con una o más variantes genéticas, asociados con CI (tabla 1)26. En el caso de la CI, todas estas variantes genéticas explican aproximadamente un 15% de la heredabilidad de este fenotipo. Algunas de estas variantes también están relacionadas con el metabolismo lipídico, la presión arterial y la inflamación, confirmando la importancia de estos factores de riesgo en la etiopatogenia de la CI27. Aproximaciones más recientes que utilizan la biología de sistemas han identificado algunos mecanismos y rutas metabólicas en las que los genes asociados con la CI están sobrerrepresentados, como el metabolismo lipídico, el de los aminoácidos que contienen sulfuro, el de las poliaminas, la inmunidad innata, la degradación de la matriz extracelular, y las proteínas mediadoras de la respuesta a colapsina28. Además, la mayoría de estas variantes se encuentran en regiones intergénicas próximas a las regiones promotoras de genes, lo que indica que pueden regular su expresión y apunta a la relevancia de la expresión génica y la epigenética en la determinación del riesgo de CI29. Todos los resultados de los estudios GWAS relacionados con la CI, sus factores de riesgo, y otros fenotipos complejos se han incluido en un catálogo para facilitar el acceso a los investigadores y clínicos25.
Tabla 1.Resumen de los resultados principales del metanálisis más reciente de los estudios de asociación de genoma completo que analizan variantes genéticas asociadas con cardiopatía isquémica*
SNP Gen próximo Cromosoma Alelo de riesgo/no riesgo Frecuencia alelo de riesgo p OR (IC95%) rs11206510 PCSK9 1 T/C 0,848 2,34 E-08 1,08 (1,05-1,11) rs9970807 PPAP2B 1 C/T 0,915 5,00 E-14 1,13 (1,10-1,17) rs7528419 SORT1 1 A/G 0,786 1,97 E-23 1,12 (1,10-1,15) rs6689306 IL6R 1 A/G 0,448 2,60 E-09 1,06 (1,04-1,08) rs67180937 MIA3 1 G/T 0,663 1,01 E-12 1,08 (1,06-1,11) rs16986953 AK097927 2 A/G 0,105 1,45 E-08 1,09 (1,06-1,12) chr2:21378433:D APOB 2 D/I 0,746 2,89 E-08 1,07 (1,04-1,10) chr2:44074126:D ABCG5-ABCG8 2 I/D 0,745 2,60 E-08 1,06 (1,04-1,09) rs7568458 VAMP5-VAMP8-GGCX 2 A/T 0,449 3,62 E-10 1,06 (1,04-1,08) rs17678683 ZEB2-ACO74093.1 2 G/T 0,088 3,00 E-09 1,10 (1,07-1,14) chr2:203828796:I WDR12 2 I/D 0,108 2,15 E-18 1,15 (1,11-1,18) chr3:138099161:I MRAS 3 I/D 0,163 2,89 E-09 1,08 (1,05-1,10) rs4593108 EDNRA 4 C/G 0,795 8,82 E-10 1,07 (1,05-1,10) rs72689147 GUCY1A3 4 G/T 0,817 6,07 E-09 1,07 (1,05-1,10) rs17087335 REST-NOA1 4 T/G 0,210 4,60 E-08 1,06 (1,04-1,09) rs273909 SLC22A4-SLC22A5 5 G/A 0,117 1,24 E-04 1,06 (1,03-1,09) rs6903956 ADTRP-C6orf105 6 A/G 0,354 0,96 1,00 (0,98-1,02) rs9349379 PHACTR1 6 G/A 0,432 1,81 E-42 1,14 (1,12-1,16) rs17609940 ANKS1A 6 G/C 0,824 0,03 1,03 (1,00-1,05) rs56336142 KCNK5 6 T/C 0,807 1,85 E-08 1,07 (1,04-1,09) rs12202017 TCF21 6 A/G 0,700 1,98 E-11 1,07 (1,05-1,09) rs55730499 SLC22A3-LPAL2-LPA 6 T/C 0,056 5,39 E-39 1,37 (1,31-1,44) rs4252185 PLG 6 C/T 0,060 1,64 E-32 1,34 (1,28-1,41) rs2107595 HDAC9 7 A/G 0,200 8,05 E-11 1,08 (1,05-1,10) rs10953541 7q22 7 C/T 0,783 1,02 E-05 1,05 (1,03-1,08) rs11556924 ZC3HC1 7 C/T 0,687 5,34 E-11 1,08 (1,05-1,10) rs17087335 NOS3 7 T/C 0,060 1,70 E-09 1,14 (1,09-1,19) rs264 LPL 8 G/A 0,853 1,06 E-05 1,06 (1,03-1,09) rs2954029 TRIB1 8 A/T 0,551 2,61E-06 1,04 (1,03-1,06) rs2891168 9p21 9 G/A 0,489 2,29 E-98 1,21 (1,19-1,24) rs2891168 9p21 9 G/A 0,489 2,29 E-98 1,21 (1,19-1,24) rs2519093 ABO 9 T/C 0,191 1,19 E-11 1,08 (1,06-1,11) rs2487928 KIAA1462 10 A/G 0,418 4,41 E-11 1,06 (1,04-1,08) rs1870634 CXCL12 10 G/T 0,637 5,55 E-15 1,08 (1,06-1,10) rs1412444 LIPA 10 T/C 0,369 5,15 E-12 1,07 (1,05-1,09) rs11191416 CYP17A1-CNNM2-NT5C2 10 T/G 0,873 4,65 E-09 1,08 (1,05-1,11) rs2128739 PDGFD 11 A/C 0,324 7,05 E-11 1,07 (1,05-1,09) rs964184 ZNF259-APOA5-APOA1 11 G/C 0,185 5,60 E-05 1,05 (1,03-1,08) rs10840293 SWAP70 11 A/G 0,550 1,38 E-08 1,06 (1,04-1,08) rs2681472 ATP2B1 12 G/A 0,201 6,17 E-11 1,08 (1,05-1,10) rs3184504 SH2B3 12 T/C 0,422 1,03 E-09 1,07 (1,04-1,09) rs1180803 KSR2 12 G/T 0,360 3,12 E-09 1,12 (1,08-1,16) rs9319428 FLT1 13 A/G 0,314 7,13 E-05 1,04 (1,02-1,06) rs11838776 COL4A1/A2 13 A/G 0,263 1,83 E-10 1,07 (1,05-1,09) rs10139550 HHIPL1 14 G/C 0,423 1,38 E-08 1,06 (1,04-1,08) rs4468572 ADAMTS7 15 C/T 0,586 4,44 E-16 1,08 (1,06-1,10) rs17514846 FURIN-FES 15 A/C 0,440 3,10 E-07 1,05 (1,03-1,07) rs56062135 SMAD3 15 C/T 0,790 4,50 E-09 1,07 (1,05-1,10) rs8042271 MFGE8-ABHD2 15 G/A 0,900 3,70 E-08 1,10 (1,06-1,14) rs216172 SMG6 17 C/G 0,350 5,07 E-07 1,05 (1,03-1,07) rs12936587 RAI1-PEMT-RASD1 17 G/A 0,611 8,24 E-04 1,03 (1,01-1,05) rs46522 UBE2Z 17 T/C 0,513 1,84 E-05 1,04 (1,02-1,06) rs7212798 BCAS3 17 C/T 0,150 1,90 E-08 1,08 (1,05-1,11) rs663129 PMAIP1-MC4R 18 A/G 0,260 3,20 E-08 1,06 (1,04-1,08) rs56289821 LDLR 19 G/A 0,900 4,44 E-15 1,14 (1,11-1,18) rs4420638 APOE-APOC1 19 G/A 0,166 7,07 E-11 1,10 (1,07-1,13) rs12976411* ZNF507-LOC400684 19 T/A 0,090 1,18 E-14 1,49 (1,35-1,67) rs28451064 KCNE2 (gene desert) 21 A/G 0,121 1,33 E-15 1,14 (1,10-1,17) rs180803 POM121L9P-ADORA2A 22 G/T 0,970 1,60 E-10 1,20 (1,13-1,27) A: adenina; C: citosina; D: deleción; G: guanina; I: inserción; IC95%: intervalo de confianza del 95%; OR: odds ratio; SNP: polimorfismo de base única (single nucleotide polymorphism); T: timina.
Las principales aportaciones de este tipo de estudios han sido la consistencia de sus resultados, la red de colaboraciones entre grupos y la puesta de los datos (crudos o agregados) a disposición de la comunidad científica como por ejemplo: el repositorio europeo de genoma-fenotipo30, la base de datos de genotipos y fenotipos americana dbGaP31 o los resultados agregados de las variantes genéticas asociadas con CI del consorcio CARDIoGRAMplusC4D32 que incluye más de 60.000 casos de CI y más de 123.000 controles.
Las principales limitaciones son que las variantes genéticas identificadas no tienen por qué ser las que se relacionen de forma causal con el fenotipo (puede estar en desequilibrio de ligamiento/asociada con la variante causal) y no aportan información sobre el mecanismo de la asociación, por lo que son necesarios estudios funcionales para identificar los mecanismos fisiopatológicos relacionados. Por otro lado, al ser estudios colaborativos, la definición de los fenotipos clínicos estudiados puede ser heterogénea entre estudios y, fundamentalmente, están diseñados para identificar variantes genéticas comunes con efectos pequeños, pero no para identificar variantes genéticas raras con efectos más grandes.
La heredabilidad todavía por descubrir es uno de los grandes retos actuales de la genética de las enfermedades complejas33. Esta heredabilidad puede estar relacionada con variantes comunes todavía no descubiertas, variantes raras con efectos mayores o similares a los de las variantes comunes, y con cambios en el ADN no relacionados con cambios en la secuencia de bases, sino con la estructura, y que puedan influir en la expresión génica (epigenética).
- 4.
Estudios de secuenciación del genoma. Clásicamente este tipo de metodología se ha utilizado para el estudio de enfermedades mono-oligogénicas con una clara agregación familiar. Los estudios de secuenciación pueden estar limitados a un gen, un panel de genes, el exoma (la parte del genoma que codifica proteínas) o todo el genoma. El genoma humano contiene unos 3.100 millones de nucleótidos, y el exoma engloba unos 30 millones de nucleótidos y unos 23.000 genes34. En esta revisión no se va a discutir la utilidad y las limitaciones de los métodos de secuenciación en el estudio de las enfermedades mono-oligogénicas35,36. En el caso de la CI, este tipo de estudios puede identificar variantes raras que en teoría tendrían un efecto mayor que el de las variantes comunes. Se ha realizado algún estudio en el que se ha secuenciado el exoma en unos 6.700 casos y 6.700 controles, y se han identificado variantes raras en los genes LDLR y APOA5 que se asocian con un mayor riesgo de infarto agudo de miocardio37 con OR que oscilan entre 1,5 y 4,5 y que apoyan de nuevo la relevancia del metabolismo lipídico (colesterol unido a lipoproteínas de baja densidad [cLDL] y triglicéridos). Otros estudios se han centrado en genes concretos y han identificado variantes raras relacionadas con la CI en genes que intervienen en el metabolismo lipídico: APOC338, NPC1L139, SCARB140, ANGPTL4, LPL y SVEP141.
La traslación del conocimiento generado sobre la arquitectura genética de la CI tiene varias aplicaciones clínicas: la identificación de nuevas dianas terapéuticas, la mejora de la estimación del riesgo cardiovascular y la farmacogenómica.
Identificación de nuevas dianas terapéuticasLa genética puede ser una herramienta muy útil en el descubrimiento o validación de dianas terapéuticas42. Clásicamente se utilizan 3 estrategias: estudios de asociación (genotipado, secuenciación), estudios de aleatorización mendeliana y estudios de secuenciación de variantes con pérdida de función.
Estudios de asociaciónLos GWAS han identificado genes que se asocian con la CI25–27. En el caso de los GWAS, únicamente un tercio de los 55 loci asociados con CI se asocian con los factores de riesgo clásicos, abriendo nuevos mecanismos y nuevas dianas terapéuticas potenciales. En un estudio reciente, en el que se analizaron los resultados de 361 GWAS publicados hasta febrero de 2011 y que identificaron 991 genes, se observó que en 63 casos los GWAS identificaron un gen asociado con una enfermedad sobre el que actúa un fármaco que se utiliza en el tratamiento/prevención de dicha patología43. Además, en 92 casos se identificaron genes asociados con un fenotipo, y sobre los que actuaban algunos fármacos que se utilizaban en el tratamiento de otra enfermedad; lo que sugiere que ese fármaco se puede reposicionar para el tratamiento de una nueva entidad.
Sin embargo, la identificación de variantes genéticas asociadas con un fenotipo no es suficiente para asumir este gen como una diana terapéutica. Como ejemplo positivo, ya en fase III de evaluación de su eficacia para la prevención de acontecimientos clínicos, están los inhibidores de la proteína PCSK9. Este gen se descubrió mediante estudios de ligamiento como asociado con la hipercolesterolemia familiar15,16. Posteriormente se identificó el mecanismo por el que la proteína PCSK9 regulaba los valores de cLDL44,45 al degradar el receptor de LDL. Después se diseñaron anticuerpos anti-PCSK9 que han demostrado su eficacia para reducir los valores de cLDL y se está a la espera de los resultados definitivos sobre su eficacia para reducir acontecimientos clínicos46,47.
Como ejemplo negativo, que demuestra la necesidad de conocer los mecanismos de la asociación entre la variante genética/gen y el fenotipo, cabe mencionar la región 9p21. Esta región fue la que se identificó como asociada con CI en los primeros GWAS publicados en 200723,24. Las variantes genéticas que se asocian con CI se encuentran en una región intergénica, próxima a un cluster de genes que regulan el ciclo celular (CDKN2A y CDKN2B) y codifican un ARN no codificante de proteína (CDKN2BAS o ANRIL). Aunque se han propuesto varias hipótesis que explicarían su asociación con la CI, el mecanismo funcional todavía no está claro48; por lo que se ve limitado el diseño de nuevos fármacos para la prevención cardiovascular49.
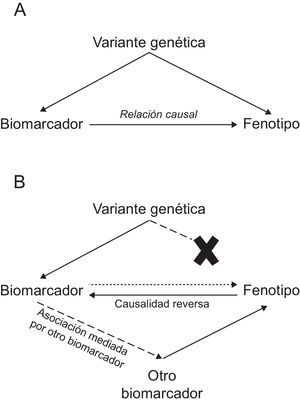
Estudios de aleatorización mendelianaEste tipo de estudios utiliza variantes genéticas como un instrumento para determinar si la relación entre un biomarcador que se asocia con la enfermedad es causal o no. La definición de causalidad es muy importante, ya que es un requisito necesario para definir si la modificación de ese biomarcador como diana terapéutica puede reducir el riesgo de una enfermedad. Los estudios de aleatorización mendeliana se fundamentan en 2 principios básicos de la biología: a) que las variantes genéticas se asignan de forma aleatoria en la meiosis, y b) que siguiendo la segunda ley de Mendel, las variantes genéticas se transmiten de una generación a otra de forma independiente entre ellas. En la práctica, estos principios implican que el grupo de individuos portadores de la variante genética que se asocia con el biomarcador es igual que el grupo de individuos no portadores de la variante genética en todo excepto en esa variante en estudio, de manera que se puede considerar un ensayo clínico aleatorizado en el que, además, la intervención ha durado toda la vida.
De manera que en este tipo de estudio se selecciona un biomarcador que se asocia con la enfermedad, una o varias variantes genéticas que se asocian con el biomarcador, y se analiza si la variante genética se asocia con la enfermedad: si existe asociación, sugiere que la relación entre biomarcador y enfermedad es causal; si no existe asociación, indica que la relación entre biomarcador y enfermedad no es causal (en este caso, la asociación entre el biomarcador y el fenotipo puede ser causalidad reversa o mediada por otro biomarcador) (figura).

Este tipo de aproximación se ha utilizado en el análisis de la relación causal de algunos biomarcadores y la CI: sugiere una relación causal con cLDL50,51, triglicéridos50–52, interleucina 653, adiposidad54 y diabetes55,56 y cuestiona la causalidad en el caso de colesterol unido a lipoproteínas de alta densidad50,51,57, proteína C-reactiva58,59, ácido úrico60, cistatina C61, adiponectina62, fospolipasa A263,64, bilirrubina65 y vitamina D (tabla 2)66. Asimismo, ha servido para determinar que las variantes genéticas del gen PCSK9 que se asocian con menores concentraciones de cLDL también se asocian con mayores valores de glucemia, mayor cintura y riesgo de diabetes, apoyando la necesidad de monitorizar los potenciales efectos adversos de los fármacos anti-PCSK967.
Resultados de los estudios de aleatorización mendeliana que han analizado la relación causal entre algunos biomarcadores y la cardiopatía isquémica
| Apoyan la relación causal con CI | Cuestionan la relación causal con CI |
|---|---|
| cLDL | cHDL |
| Triglicéridos | Proteína C reactiva |
| IL-6 | Ácido úrico |
| Adiposidad | Cistatina C |
| Diabetes | Adiponectina |
| Fosfolipasa A2 | |
| Bilirrubina | |
| Vitamina D |
cHDL: colesterol unido a lipoproteínas de alta densidad; CI: cardiopatía isquémica; cLDL: colesterol unido a lipoproteínas de baja densidad; IL-6: interleucina 6.
Un diseño que está demostrando ser muy eficaz para identificar y validar algunas proteínas como diana terapéutica es el análisis de las variantes que producen una pérdida de función de la proteína codificada, y que simularían el efecto de un fármaco que inactivara o redujera la acción de esta proteína. Normalmente estas variantes son raras, por lo que se necesitan estudios con muestras de gran tamaño y metodología de secuenciación para detectar la presencia de estas variantes en algunos genes concretos. Un ejemplo claro ha sido la validación de la proteína NPC1L1 (diana terapéutica de ezetimiba) para reducir el riesgo de presentar CI39, que se ha confirmado en el ensayo clínico IMPROVE-IT68 en pacientes en prevención secundaria. Otro ejemplo que ha permitido identificar una nueva diana terapéutica es el gen ANGPTL4. En este gen se han identificado variantes genéticas con pérdida de función de la proteína que se asocian con una reducción de los valores de triglicéridos y un menor riesgo de CI41,69 y se ha desarrollado un anticuerpo monoclonal que ha demostrado ser eficaz para reducir las concentraciones de triglicéridos en modelos animales69, aunque quedan pendientes los eudios de seguridad y eficacia en humanos.
Mejora de la estimación del riesgo cardiovascularLa Sociedad Europea de Cardiología recomienda estimar el riesgo cardiovascular para definir las estrategias preventivas más adecuadas a cada individuo según su riesgo70. Las funciones de riesgo son el instrumento recomendado para esta estimación. En España existen diferentes funciones adaptadas y desarrolladas como son el SCORE calibrado71, REGICOR72, FRESCO73 y ERICE74; aunque únicamente hay datos disponibles de la validez de 2 de ellas72,73. Estas funciones calculan la probabilidad de presentar un acontecimiento cardiovascular/coronario en los próximos 10 años según la exposición a los factores de riesgo clásicos. Una de las limitaciones de las funciones de riesgo es su baja sensibilidad, es decir, una parte importante de los acontecimientos se presentan en individuos categorizados como de riesgo bajo o moderado72. Por este motivo, se están evaluando nuevos biomarcadores que se puedan incluir en las funciones de riesgo para aumentar su sensibilidad75 (especialmente en el grupo de individuos de riesgo moderado) y para realizar esta evaluación hay que seguir unas recomendaciones76.
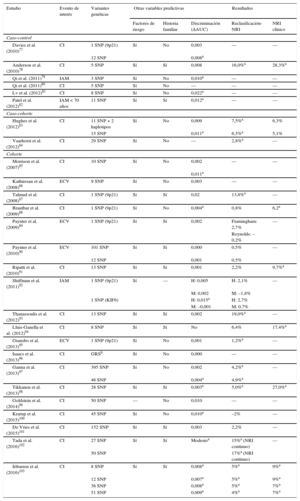
Las variantes genéticas asociadas con CI son candidatas para mejorar la capacidad predictiva de las funciones de riesgo actuales. Entre las ventajas se incluyen que se determinan una única vez en la vida; su coste es bajo, y la precisión de los métodos de genotipado es muy alta. Entre las limitaciones hay que señalar que las variantes genéticas individuales tienen una magnitud de asociación pequeña (OR = 1,1 a 1,4) que limitan su capacidad predictiva, aunque esta magnitud aumenta cuando se consideran varias variantes que se incluyen en una puntuación de carga genética de riesgo (genetic risk score) y es similar a la de los factores de riesgo clásicos. Son varios los estudios que han evaluado el valor añadido de incluir la información genética en las funciones de riesgo clásicas77–103. En casi todos ellos se observa una asociación entre las variantes genéticas y el riesgo de presentar CI, independientemente de los factores de riesgo cardiovascular clásicos y de los antecedentes familiares de CI. La mayoría de los estudios no encuentran una mejora en la capacidad de discriminación al incluir la información genética en la función; aunque sí que observan una mejora en la reclasificación de los individuos en las categorías de riesgo, especialmente en aquellos con riesgo moderado (tabla 3). En un metanálisis reciente también se ha observado que la carga genética de riesgo se asocia con un peor pronóstico en aquellos pacientes que ya presentaban la enfermedad. Además, la eficacia de las estatinas era mayor en aquellos pacientes con carga genética de riesgo más elevada tanto en prevención primaria como en secundaria104.
Características y resultados más importantes de los estudios que han analizado el valor añadido de la información genética en la estimación del riesgo cardiovascular
| Estudio | Evento de interés | Variantes genéticas | Otras variables predictivas | Resultados | |||
|---|---|---|---|---|---|---|---|
| Factores de riesgo | Historia familiar | Discriminación (ΔAUC) | Reclasificación-NRI | NRI clínico | |||
| Caso-control | |||||||
| Davies et al. (2010)77 | CI | 1 SNP (9p21) | Sí | No | 0,003 | — | — |
| 12 SNP | 0,008a | ||||||
| Anderson et al. (2010)78 | CI | 5 SNP | Sí | Sí | 0,008 | 16,0%a | 28,3%a |
| Qi et al. (2011)79 | IAM | 3 SNP | Sí | No | 0,010a | — | — |
| Qi et al. (2011)80 | CI | 5 SNP | Sí | No | — | — | — |
| Lv et al. (2012)81 | CI | 8 SNP | Sí | No | 0,022a | — | — |
| Patel et al. (2012)82 | IAM < 70 años | 11 SNP | Sí | Sí | 0,012a | — | — |
| Caso-cohorte | |||||||
| Hughes et al. (2012)83 | CI | 11 SNP + 2 haplotipos | Sí | No | 0,009 | 7,5%a | 6,3% |
| 15 SNP | 0,011a | 6,5%a | 5,1% | ||||
| Vaarhorst et al. (2012)84 | CI | 29 SNP | Sí | No | — | 2,8%a | — |
| Cohorte | |||||||
| Morrison et al. (2007)85 | CI | 10 SNP | Sí | No | 0,002 | — | — |
| 0,011a | |||||||
| Kathiresan et al. (2008)86 | ECV | 9 SNP | Sí | No | 0,003 | — | — |
| Talmud et al. (2008)87 | CI | 1 SNP (9p21) | Sí | Sí | 0,02 | 13,8%a | — |
| Brautbar et al. (2009)88 | CI | 1 SNP (9p21) | Sí | No | 0,004a | 0,8% | 6,2a |
| Paynter et al. (2009)89 | ECV | 1 SNP (9p21) | Sí | Sí | 0,002 | Framingham: 2,7% | — |
| Reynolds: –0,2% | |||||||
| Paynter et al. (2010)90 | ECV | 101 SNP | Sí | Sí | 0,000 | 0,5% | — |
| 12 SNP | 0,001 | 0,5% | |||||
| Ripatti et al. (2010)91 | CI | 13 SNP | Sí | Sí | 0,001 | 2,2% | 9,7%a |
| Shiffman et al. (2011)92 | IAM | 1 SNP (9p21) | Sí | — | H: 0,005 | H: 2,1% | — |
| M: 0,002 | M: –1,8% | ||||||
| 1 SNP (KIF6) | H: 0,015a | H: 2,7% | |||||
| M: –0,001 | M: 0,7% | ||||||
| Thanassoulis et al. (2012)93 | CI | 13 SNP | Sí | Sí | 0,002 | 19,0%a | — |
| Lluis-Ganella et al. (2012)94 | CI | 8 SNP | Sí | Sí | No | 6,4% | 17,4%a |
| Gransbo et al. (2013)95 | ECV | 1 SNP (9p21) | Sí | No | 0,001 | 1,2%a | — |
| Isaacs et al. (2013)96 | CI | GRSb | Sí | No | 0,000 | — | — |
| Ganna et al. (2013)97 | CI | 395 SNP | Sí | No | 0,002 | 4,2%a | — |
| 46 SNP | 0,004a | 4,9%a | |||||
| Tikkanen et al. (2013)98 | CI | 28 SNP | Sí | Sí | 0,003a | 5,0%a | 27,0%a |
| Goldstein et al. (2014)99 | CI | 50 SNP | — | No | 0,010 | — | — |
| Krarup et al. (2015)100 | CI | 45 SNP | Sí | No | 0,010a | –2% | — |
| De Vries et al. (2015)101 | CI | 152 SNP | Sí | Sí | 0,003 | 2,2% | — |
| Tada et al. (2016)102 | CI | 27 SNP | Sí | Sí | Modestoa | 15%a (NRI continuo) | — |
| 50 SNP | 17%a (NRI continuo) | ||||||
| Iribarren et al. (2016)103 | CI | 8 SNP | Sí | Sí | 0,008a | 5%a | 9%a |
| 12 SNP | 0,007a | 5%a | 9%a | ||||
| 36 SNP | 0,008a | 5%a | 7%a | ||||
| 51 SNP | 0,009a | 4%a | 7%a | ||||
ΔAUC: cambio en el área bajo la curva o estadístico C; CI: cardiopatía isquémica; ECV: enfermedad cardiovascular; GRS: carga genética de riesgo (genetic risk score); H: hombres; IAM: infarto agudo de miocardio; M: mujeres; NRI: índice de reclasificación (net reclassification index); SNP: polimorfismo de base única (single nucleotide polymorphism).
La variabilidad interindividual en la respuesta a un fármaco también puede estar relacionada con la variabilidad genética105,106. En el área cardiovascular hay 2 fármacos que pueden servir de ejemplo: las estatinas y el clopidogrel. En el caso de las estatinas se ha identificado que la variabilidad genética en algunos genes (SORT1/CELSR2/PSRC1, SLCO1B1, APOE y LPA) puede explicar un 5% de la variabilidad interindividual en el descenso de la concentración de cLDL107 y la variabilidad genética en el gen SLCO1B1 también se asocia de forma consistente con un mayor riesgo de miopatía inducida por simvastatina108.
El clopidogrel es un fármaco que se metaboliza por el sistema del citocromo P450 (CYP) del hígado para convertirse en un metabolito activo que se une al receptor P2Y12 de la plaqueta para su acción antiagregante. En este proceso interviene el CYP2C19 y se han descrito variantes genéticas en este gen, algunas aumentan y otras disminuyen la actividad de este CYP, que podrían estar relacionadas con acontecimientos clínicos como la trombosis del stent, el riesgo de sangrado o la mortalidad cardiovascular. En 2010 la Food and Drug Administration de Estados Unidos añadió una advertencia en el prospecto del clopidogrel advirtiendo que algunos pacientes, y según sus características genéticas, podían no obtener los beneficios esperados del fármaco109. Aunque las sociedades científicas norteamericanas publicaron un artículo de consenso en el que concluían que la utilidad de las pruebas genéticas para determinar el metabolismo del clopidogrel estaba pendiente de definir110, otras sociedades aconsejan su uso111. Un metanálisis reciente también concluye que la evidencia actual no es suficiente para individualizar la utilización de este fármaco guiada por el genotipo CYP2C19112.
ALGUNOS RETOS PARA EL FUTUROEn los últimos años se ha avanzado mucho en el conocimiento de las bases genéticas de la CI; lo que ha contribuido a entender mejor los mecanismos etiopatogénicos que determinan la progresión y aparición de esta enfermedad, a identificar nuevas dianas terapéuticas y a mejorar la estimación del riesgo cardiovascular113. De todos modos, todavía queda mucho camino por recorrer:
- •
La heredabilidad explicada es pequeña y queda por descubrir el papel de las variantes raras con efectos modestos; de las modificaciones epigenéticas (metilación, histonas y ARN no codificantes), y de la interacción gen-ambiente y gen-gen en la determinación de la susceptibilidad individual a presentar CI.
- •
Un reto importante para los próximos años será la integración de la información que provenga de múltiples «ómicas» (genómica, epigenómica, transcriptómica, proteómica, metabolómica) y su correlación con el fenotipo (imagen, funcional, clínico). La interacción entre todas estas capas de información es la que determina el funcionalismo celular de los órganos y sistemas. En este punto, un aspecto importante que cabe señalar es que esta interacción puede ser específica y diferencial en líneas celulares, órganos y sistemas; lo que añade complejidad a su estudio. El desarrollo de nuevas técnicas de análisis de grandes bases de datos y la biología de sistemas serán elementos fundamentales para continuar avanzando.
- •
Toda esta información puede contribuir a una mayor precisión en la definición del fenotipo y es probable que en el futuro no se hable de CI, sino de diferentes subgrupos de entidades dentro de esta patología. Esta nueva definición fenotípica y molecular contribuirá a catalizar el desarrollo de la medicina de precisión.
Este trabajo se ha financiado parcialmente con ayudas del Instituto de Salud Carlos III-FEDER (FIS PI15/00051; Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares) y la Agència de Gestió d’Ajuts Universitaris i de Recerca de la Generalitat de Catalunya (2014SGR240). S. Sayols-Baixeras recibe una ayuda iPFIS del Instituto de Salud Carlos III-FEDER (IFI14/00007).
CONFLICTO DE INTERESESR. Elosua es miembro del comité asesor de Gendiag e inventor, en una patente, de una prueba genética para la estimación del riesgo cardiovascular cuyo titular es Gendiag.exe.