


Son escasas las publicaciones sobre aparición de hipertensión arterial pulmonar tras la cirugía de switch arterial en periodo neonatal para la corrección de la transposición de grandes arterias. Se evalúa la frecuencia y el comportamiento clínico de esta complicación en una serie de pacientes.
MétodosSe revisó la base de datos y se seleccionó a pacientes con transposición de grandes vasos corregida con switch arterial neonatal en el centro en los que con el tiempo apareció hipertensión pulmonar.
ResultadosSe halló a 2 pacientes (1,3%) con transposición de grandes arterias corregida con éxito en la primera semana de vida que luego presentaron hipertensión arterial pulmonar. El primero es una niña de 7 años con diagnóstico de hipertensión pulmonar grave a los 8 meses de edad, sin respuesta a tratamiento médico, que precisó trasplante pulmonar. La anatomía patológica mostró hallazgos compatibles con hipertensión arterial pulmonar grave. El segundo es un niño de 24 meses con diagnóstico de hipertensión pulmonar grave a los 13 meses, sin respuesta al tratamiento médico.
ConclusionesLa hipertensión arterial pulmonar es una complicación infrecuente pero muy grave cuya aparición se debe investigar en todo paciente con transposición de grandes vasos sometido a operación de switch arterial neonatal con el fin de instaurar un tratamiento agresivo temprano para los pacientes afectados, dados la escasa respuesta al tratamiento y el mal pronóstico que supone.
Palabras clave
La aparición de hipertensión arterial pulmonar (HAP) en pacientes con d-transposición de grandes arterias (d-TGA) es una complicación potencialmente grave descrita hace décadas1–3. Afecta habitualmente a pacientes con d-TGA no corregida quirúrgicamente1, especialmente aquellos con defecto septal ventricular y/o ductus arterioso asociado, en los que la exposición continua a un flujo pulmonar aumentado ocasionaba finalmente un remodelado vascular y el consiguiente aumento de las resistencias vasculares pulmonares4. Tras la introducción de la cirugía de switch auricular, la incidencia de esta complicación se redujo de manera considerable5 y, tras el establecimiento de la operación de switch arterial (SA) como método de corrección quirúrgica de elección desde los años 80, los casos de HAP en pacientes con d-TGA se han comunicado muy esporádicamente6–9. Este tipo de HAP que recurre o aparece meses o años después del SA en ausencia de lesiones residuales significativas tiene un mecanismo fisiopatológico desconocido y un curso agresivo6–9, y en algunos estudios es la causa más común de mortalidad tardía tras este tipo de cirugía10.
Los objetivos de este estudio son evaluar la incidencia de HAP tras la corrección quirúrgica de la d-TGA mediante SA en periodo neonatal en una serie de pacientes y describir las características clínicas y hemodinámicas de 2 casos de HAP tras la cirugía de SA.
MÉTODOSEstudio retrospectivo realizado en un centro terciario de referencia para la cirugía cardiaca de cardiopatías congénitas, entre enero de 1995 y diciembre de 2014. Utilizando la base de datos, se recogió la información de todos los pacientes con formas simples o complejas de d-TGA a los que se realizó SA en periodo neonatal, considerando como tal las primeras 6 semanas de vida. Se excluyó a los pacientes con d-TGA compleja a los que se realizó un procedimiento quirúrgico diferente del SA. Tras la intervención quirúrgica correctora, se dio seguimiento a todos los pacientes en las consultas externas del centro incluyendo historia clínica, exploración física, electrocardiograma y ecocardiograma transtorácico. En los casos en que se sospechaba hipertensión pulmonar, se realizó cateterismo cardiaco. El diagnóstico y el tratamiento se realizaron de acuerdo con las guías clínicas vigentes11,12.
RESULTADOSLa población de estudio está constituida por 152 pacientes con d-TGA corregida en periodo neonatal mediante SA: 96 pacientes (63,2%) con d-TGA con septo interventricular íntegro y 56 (36,8%) con formas complejas de d-TGA (con defecto septal ventricular y/o coartación de aorta asociados). Tras una media de 8,5 ± 4,7 años (intervalo, 10 meses-19 años) desde la cirugía, 2 pacientes (1,3%) presentaron HAP. La tabla 1 muestra las características de los pacientes afectados y su curso clínico.
Características de los pacientes que contraen hipertensión arterial pulmonar tras la cirugía de switch arterial en periodo neonatal, evolución y resultados
| Paciente | A | B |
|---|---|---|
| Tipo de TGA | TGA con septo íntegro | TGA + DSV |
| Edad al SA (días) | 6 | 7 |
| Procedimiento quirúrgico | SA | SA + cierre de DSV |
| Edad al diagnóstico de HAP (meses) | 7 | 13 |
| Cateterismo cardiaco | PAP, 74/7 (39) mmHg PAPm/PAom, 0,96 RVP, 14 UW/m2 | PAP, 85/6 (41) mmHg PAPm/PAom, 0,78 RVP, 15 UW/m2 |
| Tratamiento | Bosentán + sildenafilo + iloprost inhalado | Bosentán + sildenafilo |
| Evolución | Trasplante pulmonar a los 3 años y 10 meses de edad | Clínicamente estable a los 24 meses de edad; clase funcional II de la NYHA |
DSV: defecto septal ventricular; HAP: hipertensión arterial pulmonar; NYHA: New York Heart Association; PAom: presión media en la aorta; PAP: presión arterial pulmonar; PAPm: presión media en la arteria pulmonar; RVP: resistencias vasculares pulmonares; SA: switch arterial; TGA: transposición de grandes arterias.
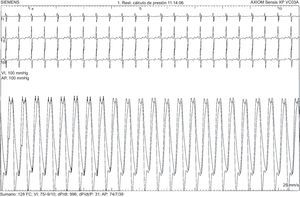
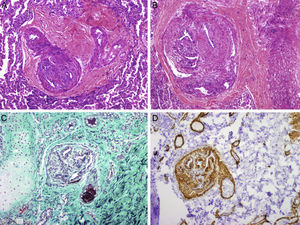
El primer paciente es una niña de 7 años con diagnóstico prenatal de d-TGA con septo interventricular íntegro a la que se realizó cirugía tipo SA a los 6 días de vida, sin complicaciones posoperatorias ni lesiones residuales. Tras una evolución inicial favorable, a los 7 meses de edad se observó estancamiento ponderal y saturación de oxígeno del 90%. La ecocardiografía mostró una comunicación interauricular pequeña con cortocircuito bidireccional y signos de hipertensión pulmonar. El cateterismo cardiaco confirmó la presión pulmonar sistémica con resistencias vasculares pulmonares elevadas y test vasodilatador pulmonar positivo (figura 1). No se encontraron otras anomalías que justificasen el cuadro clínico. Dado que la paciente tenía menos de 1 año, lo que constituye una contraindicación relativa para el uso de antagonistas del calcio, unido al perfil hemodinámico agresivo de la hipertensión pulmonar, se decidió iniciar tratamiento combinado con sildenafilo y bosentán. Tras 6 meses de tratamiento, se añadió iloprost inhalado, a pesar de lo cual la situación clínica empeoró progresivamente, y a los 3 años y 10 meses de edad, se le realizó un trasplante pulmonar. El examen anatomopatológico del pulmón mostró hallazgos compatibles con HAP y daño tisular irreversible (figura 2). A los 4 años del trasplante pulmonar, la paciente se encontraba clínicamente estable.

. C: se observa, además, fibrosis concéntrica de la pared en la arteriola de la derecha; imágenes de lesiones plexiformes con proliferación de células endoteliales, de músculo liso y miofibroblastos que forman luces secundarias intramurales, especialmente en las zonas de bifurcación de los vasos (B: hematoxilina-eosina; D: actina de músculo liso) (cortesía del Dr. J.C. Ferreres, Hospital Vall d’Hebron, Barcelona).")
Estudio anatomopatológico pulmonar. Imágenes demostrativas de vasculopatía obstructiva pulmonar: hiperplasia intimal en una arteria muscular con colagenización de la íntima y reducción del diámetro de la luz (A: hematoxilina-eosina; C: tricrómico de Masson). C: se observa, además, fibrosis concéntrica de la pared en la arteriola de la derecha; imágenes de lesiones plexiformes con proliferación de células endoteliales, de músculo liso y miofibroblastos que forman luces secundarias intramurales, especialmente en las zonas de bifurcación de los vasos (B: hematoxilina-eosina; D: actina de músculo liso) (cortesía del Dr. J.C. Ferreres, Hospital Vall d’Hebron, Barcelona).
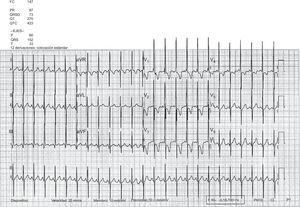
El segundo paciente es un niño de 2 años con diagnóstico prenatal de d-TGA con defecto septal ventricular asociado al que se realizó SA y cierre quirúrgico del defecto septal ventricular a los 7 días de vida, sin complicaciones ni lesiones residuales posquirúrgicas. Se mantuvo asintomático hasta los 13 meses de edad, cuando comenzó con disnea progresiva. El electrocardiograma mostró signos de crecimiento significativo de cavidades derechas con patrón de tensión (figura 3). El ecocardiograma transtorácico mostró hipertensión pulmonar grave, con gran dilatación del ventrículo derecho y compresión del ventrículo izquierdo (figura 4). El cateterismo cardiaco demostró presión pulmonar y resistencias vasculares pulmonares elevadas con test vasodilatador pulmonar negativo. Tras el inicio del tratamiento con sildenafilo y bosentán, mejoró su situación clínica. En el momento de redactar este trabajo, se mantenía en clase funcional II, aunque persistían signos ecocardiográficos de HAP grave.


Ecocardiograma transtorácico. A: proyección subcostal que muestra la gran dilatación e hipertrofia del ventrículo derecho y la compresión del ventrículo izquierdo que ocasiona. B: proyección apical de cuatro cámaras; marcada dilatación de cavidades derechas. VD: ventrículo derecho; VI: ventrículo izquierdo.
Las primeros casos de HAP asociados a d-TGA aparecen recogidos en varias series necrópsicas de pacientes no operados, con más frecuencia cuanto mayor es la edad y en aquellos con flujo pulmonar elevado1–4, hallazgos similares a los de un análisis conjunto de varias series4 que mostró una frecuencia de vasculopatía pulmonar hipertensiva de grado ≥ 3 de Heath y Edwards13 creciente con la edad, que llegaba hasta el 34% en pacientes de edad > 12 meses. Asimismo se han descrito casos de hipertensión pulmonar grave persistente en recién nacidos con TGA14,15, lo que indica que en esta cardiopatía la progresión de la vasculopatía pulmonar es temprana y rápida.
Tras la introducción del switch auricular, que se realizaba a una media de edad de 5-22,1 meses, la incidencia de esta complicación disminuyó a cifras entre el 3,5 y el 19%5,6,16,17, y tras la instauración del SA neonatal como técnica quirúrgica correctora de elección, la posterior aparición de HAP se ha publicado mucho más excepcionalmente6–9, lo que indica que la posibilidad de que aparezca vasculopatía pulmonar hipertensiva es mayor cuanto mayor sea el tiempo de exposición a la anomalía circulatoria de la transposición de grandes vasos. La frecuencia de la HAP tras el SA se encuentra alrededor del 1%9,18, similar al resultado obtenido del 1,3%, y afecta incluso a pacientes sin larga exposición prequirúrgica a un flujo pulmonar aumentado, es decir, sin larga exposición posnatal a la fisiología de la transposición, y sin lesiones residuales posquirúrgicas6,8,9, al igual que los 2 pacientes de la presente serie, en los que la cirugía se realizó precozmente. Estos hallazgos indican, por un lado, que la progresión de la HAP es más rápida en la d-TGA que en otras cardiopatías congénitas y, por otro, que la enfermedad vascular pulmonar en algunos casos aparece muy pronto, posiblemente durante el periodo fetal. Se han propuesto varias hipótesis con el fin de explicar el desarrollo acelerado de HAP en los pacientes con d-TGA. La más aceptada propone que la vasoconstricción pulmonar secundaria a la hipoxemia, el aumento del flujo pulmonar y la llegada de sangre poco oxigenada de la circulación colateral broncopulmonar serían los principales desencadenantes19. Sin embargo, esta teoría no justifica la HAP de los pacientes operados con SA neonatal, para lo cual se ha propuesto que la alteración de los patrones de flujo en la circulación fetal sería el principal desencadenante. La llegada de sangre rica en oxígeno a la arteria pulmonar y al ductus arterioso desde la vena umbilical a través de la cava inferior causaría constricción ductal y, por lo tanto, la llegada a la circulación pulmonar de un mayor volumen de sangre a mayor presión16. Este factor prenatal también aparece en la clasificación de la hipertensión pulmonar pediátrica del grupo de Panamá20, que incluye la HAP de los pacientes con d-TGA en el grupo de enfermedad vascular pulmonar prenatal o del desarrollo. En este centro no se hace sistemáticamente estudio prenatal del carácter restrictivo del foramen oval y la constricción del ductus arterioso, debido a que los distintos parámetros ecocardiográficos propuestos hasta la fecha no presentan buen poder predictivo21,22.
Aunque el mecanismo fisiopatológico que ocasiona la alteración vascular pulmonar es desconocido, resulta evidente que los pacientes con d-TGA son propensos a la HAP, probablemente en relación con un genotipo permisivo23, y el riesgo no se reduce tras la corrección quirúrgica. Por este motivo, es complicado clasificar este tipo de HAP que, según la clasificación de Niza24, podría incluirse en el grupo de HAP posoperatoria o en el de HAP coincidente con cardiopatía congénita, que incluye a pacientes con HAP de comportamiento similar al de la HAP idiopática, escasa respuesta al tratamiento médico y mal pronóstico, cuya incidencia en la población general es de 1-2 casos/millón de habitantes/año25 y que ha sido la norma en los pacientes con d-TGA publicados hasta el momento6–9,26 (tabla 2).
Pacientes con hipertensión arterial pulmonar tras corrección de transposición de grandes arterias mediante switch arterial
| Autores | Edad a la cirugía | Edad al diagnóstico de HAP | Evolución |
|---|---|---|---|
| Sreeram et al7 | 4 meses 1,5 meses 6 meses | 7 meses 2 años 8 meses | Muerte a los 8 meses Muerte a los 2 años Estable; HAP grave |
| Rivenes et al9 | 4 días | 42 meses | Pendiente de trasplante pulmonar |
| Cordina et al6 | < 1 mes | 16 años | Estable; clase funcional II de la NYHA |
| Torres et al8 | 11 días | 9 años | Estable; HAP moderada |
| Watanabe et al26 | 6 meses | 7 años | Trasplante pulmonar (23 años) |
| Este estudio | 6 días 7 días | 7 meses 13 meses | Trasplante pulmonar (3 años) Estable; HAP grave |
HAP: hipertensión arterial pulmonar; NYHA: New York Heart Association.
El intervalo posoperatorio hasta el diagnóstico de HAP varía entre 7 meses y 16 años6,7. En este caso, los 2 pacientes afectados se encuentran entre los más jóvenes publicados hasta este momento, con diagnóstico de HAP a los 7 y 13 meses, y en ellos la evolución también ha sido agresiva. El primero con mala respuesta al tratamiento médico y trasplante pulmonar a los 3 años, con lesiones anatomopatológicas de grado 4 de Heath y Edwards, y el segundo con evidencia ecocardiográfica de HAP grave a pesar del tratamiento vasodilatador pulmonar.
CONCLUSIONESLa aparición de HAP tras la corrección quirúrgica mediante SA en pacientes con d-TGA es una complicación infrecuente pero potencialmente muy grave que presenta características similares a la HAP idiopática y tiene gran impacto en la morbimortalidad de los pacientes afectados. Es fundamental dar estrecho seguimiento a todos los pacientes a los que se realizó SA en periodo neonatal, con el fin de detectar precozmente la presencia de HAP y establecer medidas terapéuticas agresivas desde el primer momento, dada la alta probabilidad de evolución rápida y agresiva.
CONFLICTO DE INTERESESNinguno.
- –
La aparición de HAP asociada a la transposición de grandes arterias es una complicación grave cuya frecuencia ha ido en descenso con la introducción de nuevas y más precoces técnicas quirúrgicas, como el SA. Sin embargo, la etiopatogenia y la incidencia de aparición de HAP tras este tipo de corrección quirúrgica son desconocidas.
- –
La incidencia a medio plazo de HAP tras cirugía de SA es del 1,3%. El perfil hemodinámico y clínico es similar al de la HAP idiopática, de comportamiento clínico agresivo y pronóstico grave, por lo que son precisos un diagnóstico temprano y un tratamiento intensivo precoz.