

La hipertensión arterial pulmonar (HAP) se caracteriza por aumento de resistencias vasculares pulmonares, disfunción progresiva del ventrículo derecho y muerte. A pesar de los avances, sigue asociada a alta morbimortalidad. El objetivo del estudio es describir el tratamiento de esta enfermedad y determinar factores pronósticos de pacientes con HAP tratados en un centro de referencia nacional a lo largo de 30 años.
MétodosSe estudió a 379 pacientes consecutivos diagnosticados de HAP (enero de 1984-diciembre de 2014). Se los distribuyó en 3 intervalos de tiempo: previo a 2004, 2004-2009 y 2010-2014, y se analizaron los factores pronósticos de deterioro clínico.
ResultadosLa mediana de edad de los pacientes es 44 años (el 68,6% eran mujeres) y estaban en clase funcional III-IV el 72%. Se observó un incremento en etiologías más complejas: enfermedad venooclusiva e hipertensión portopulmonar en el último periodo. La terapia combinada de inicio aumentó (el 5% previo a 2004 frente al 27% posterior a 2010; p < 0,05). El análisis multivariable mostró como factores independientes de deterioro clínico edad, sexo, etiología y variables combinadas (p < 0,05). La supervivencia libre de muerte o trasplante al primero, el tercero y el quinto año fueron del 92,2, el 80,6 y el 68,5% respectivamente. La mediana de supervivencia fue 9 años (intervalo de confianza del 95%, 7,532-11,959).
ConclusionesLa HAP es una enfermedad heterogénea y compleja. La mediana de supervivencia libre de muerte o trasplante en nuestra serie es 9 años. La estructura de una unidad multidisciplinaria de HAP debe adaptarse con rapidez a los cambios que se producen en el tiempo incorporando nuevas técnicas diagnósticas y terapéuticas.
Palabras clave
Vídeo del artículo

La hipertensión arterial pulmonar (HAP) se define como un grupo de enfermedades caracterizadas por aumento progresivo de la resistencia vascular pulmonar que conduce a insuficiencia ventricular derecha y muerte. El pronóstico se condiciona por las interacciones fisiopatológicas entre la tasa de progresión de los cambios obstructivos en la microcirculación pulmonar y la respuesta adaptativa del ventrículo derecho. Los principales factores pronósticos conocidos en esta enfermedad son la expresión de la disfunción ventricular derecha.
La HAP es una enfermedad rara con una prevalencia estimada en distintos registros entre 15 y 26 casos/millón habitantes mayores de 14 años1. Ha sido necesario un esfuerzo de la comunidad científica para diseñar y realizar más de 30 ensayos clínicos multicéntricos2, que han permitido el desarrollo de 5 familias de fármacos: análogos de prostaciclinas, inhibidores de la fosfodiesterasa 5, estimuladores de gualinato ciclasa, agonistas de los receptores de prostaciclinas y antagonistas de los receptores de endotelina. Los fármacos y las novedades en el diagnóstico y el pronóstico han revolucionado el tratamiento de la HAP, que se ha plasmado en 3 guías de práctica clínica que engloban la evidencia científica acumulada en 2004, 2009 y 2015 respectivamente3–5.
Inicialmente se consideraba que el trasplante pulmonar (TP) era el único tratamiento disponible. Tras el descubrimiento de distintos fármacos específicos, el TP es la opción terapéutica final para pacientes que no responden al tratamiento farmacológico3. A pesar de los avances, la HAP sigue siendo una enfermedad asociada a alta morbimortalidad, con una supervivencia en nuestro medio del 65% a 5 años1.
El objetivo de nuestro estudio es describir la evolución de las estrategias terapéuticas y determinar los factores pronósticos y la supervivencia a largo plazo de una cohorte de pacientes diagnosticados de HAP, en un centro de referencia nacional durante 3 décadas.
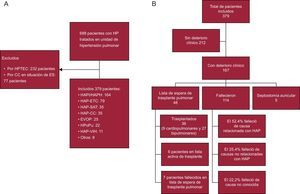
MÉTODOSDiseño del estudio y poblaciónEstudio observacional ambispectivo de una cohorte de pacientes diagnosticados de HAP del grupo 1 (HAP idiopática, familiar o heredable, formas asociadas a enfermedades del tejido conectivo, virus de la inmunodeficiencia humana [VIH], hipertensión portal, aceite de colza, cardiopatías congénitas, enfermedad venooclusiva pulmonar [EVOP] y otras etiologías menos frecuentes [Rendu Osler y anemia hemolítica] desde enero de 1984 hasta diciembre de 2014, atendidos en la Unidad Multidisciplinar de Hipertensión Pulmonar del Hospital Universitario 12 de Octubre. El estudio fue aprobado por el Comité Ético del Hospital Universitario 12 de Octubre. La inclusión de pacientes en la base de datos ha sido prospectiva desde el año 2000 y retrospectiva previamente. Se estableció como fecha de diagnóstico el primer cateterismo cardiaco derecho realizado. Se excluyeron cardiopatías congénitas en situación de Eisenmenger e hipertensión pulmonar tromboembólica crónica (figura 1).

Diagrama de flujo de los pacientes y de deterioro clínico. A: distribución de los pacientes. B: distribución del deterioro clínico. CC: cardiopatías congénitas; ES: síndrome de Eisenmenger; ETC: enfermedad del tejido conectivo; EVOP: enfermedad venooclusiva pulmonar; HAP: hipertensión arterial pulmonar; HAPH: HAP heredable; HAPI: HAP idiopática; HP: hipertensión pulmonar; HPoPu: hipertensión portopulmonar; HPTEC: hipertensión pulmonar tromboembólica crónica; SAT: síndrome de aceite tóxico; VIH: virus de la inmunodeficiencia humana.
Se ha distribuido a los pacientes en 3 intervalos de tiempo, en función de las guías disponibles en cada periodo (guías de la Sociedad Europea de Cardiología/European Respiratory Society):
- •
Diagnóstico previo al 1 de enero de 2004.
- •
Del 1 de enero de 2004 al 31 de diciembre de 2009 (guías 2004)4.
- •
Del 1 de enero de 2010 al 31 de diciembre de 2014 (guías 2009)5.
El diagnóstico de HAP se basó en el uso del algoritmo diagnóstico y criterios hemodinámicos reflejado en las guías en cada momento.
Las variables analizadas al diagnóstico fueron: datos demográficos, etiología de HAP, clase funcional (CF), prueba de marcha de 6 minutos (PM6M), presión auricular derecha, gasto cardiaco, presión media arteriopulmonar, resistencias vasculares pulmonares, derrame pericárdico y tratamiento inicial. El evento «deterioro clínico» se definió con el primer evento ocurrido: fallecimiento, inclusión en lista de espera de TP o septostomía auricular.
Análisis estadísticoLos resultados descriptivos se presentan con frecuencia y porcentaje en las variables cualitativas y con medias ± desviación estándar o mediana [intervalo intercuartílico] en las cuantitativas. La significación estadística (p < 0,05), de comparaciones entre grupos de variables cualitativas, se calculó mediante test exacto de Fisher, y en variables cuantitativas con distribución normal, se aplicó la prueba de la t de Student y análisis de la varianza para grupos independientes, según se compararan 2 o más grupos. Se utilizaron pruebas no paramétricas cuando la variable no seguía una distribución normal.
Para identificar parámetros «predictores de deterioro clínico», se realizó el análisis bivariable entre cada variable respecto al tiempo hasta el deterioro, mediante curvas de Kaplan-Meier y el log-rank test para comparar las curvas. Posteriormente se seleccionaron aquellas con significación estadística (p < 0,05) en los análisis bivariables y se incluyeron en el modelo de regresión multivariable de Cox del tiempo hasta el deterioro.
Para analizar si la etiología era factor pronóstico, se agruparon en función de la mediana de supervivencia obtenida en las curvas de Kaplan-Meier y sin que mostrasen diferencias significativas entre ellas para conformar un grupo. Un primer grupo se formó por HAP idiopática/HAP heredable/HAP-aceite de colza/HAP-cardiopatías congénitas e HAP-VIH (mediana de supervivencia, 11,9 años; intervalo de confianza del 95% [IC95%], 8,4-18,1); el segundo se formó por HAP-enfermedad del tejido conectivo/hipertensión portopulmonar/otras (mediana de supervivencia, 5,9 años; IC95%, 4,4-8,1), y finalmente EVOP en el grupo 3 (mediana de supervivencia, 2,5 años; IC95%, 1,2-7,1). Las gráficas de Kaplan-Meier sobre este análisis están disponibles en la figura del .
Basándose en la recomendación de las últimas guías de práctica clínica6, se establecieron criterios compuestos como predictores de deterioro clínico combinando la CF, la distancia en la PM6M y/o la presión auricular derecha. Se definen así los siguientes criterios: criterio 1, CF III o CF IV con PM6M < 475 m; criterio 2, CF III o CF IV con presión auricular derecha > 8 mmHg. Finalmente, se seleccionaron como factores pronósticos las variables con p < 0,05 en el modelo multivariable. El resultado final muestra el riesgo relativo (hazard ratio) y el IC95%. Para calcular sobre el total de la muestra la proporción de concordancia entre la predicción del modelo multivariable y el resultado observado, se utilizó el método C–index.
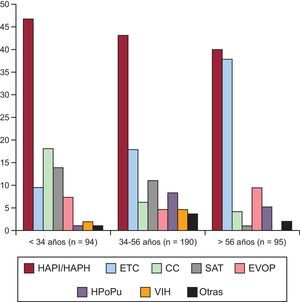
RESULTADOSDel total de 688 pacientes evaluados en nuestra unidad, se analizó a 379 que cumplían los criterios de inclusión (figura 1A). La distribución del deterioro clínico se muestra en la figura 1B. Las características principales al diagnóstico se muestran en la tabla 1. En la tabla 2 se exponen las diferencias por intervalos de tiempo. La distribución de etiologías según la época de diagnóstico muestra una presencia constante de HAP idiopática/heredable a lo largo del tiempo, y se observa una concentración de HAP-aceite de colza previa a 2004 y un creciente aumento de EVOP, HAP-cardiopatías congénitas e hipertensión portopulmonar a lo largo del tiempo. La distribución de etiologías según percentil de edad (figura 2) muestra un descenso de HAP idiopática/heredable, HAP-cardiopatías congénitas y HAP-aceite de colza conforme se incrementa la edad. En mayores de 56 años, se advierte un aumento de formas asociadas a enfermedad del tejido conectivo (el 92% esclerodermia). A partir de 2011 se empezó a realizar estudio genético, y se detectaron mutaciones genéticas en un 65% de formas familiares de la enfermedad y un 18% de casos de HAP idiopática esporádica. Todos los pacientes con EVOP familiar presentaron mutación en el gen eukaryotic initiation factor-2 kinases A4 (EIF2AK4).
Características basales
| Pacientes, n | 379 |
| Sexo | |
| Mujer | 260 (69) |
| Clasificacion clínica de HAP | |
| Cardiopatía congénita | 33 (9) |
| Enfermedad del tejido conectivo | 79 (21) |
| EVOP | 25 (7) |
| Hipertensión portopulmonar | 22 (6) |
| Idiopática | 164 (43) |
| Aceite tóxico | 35 (9) |
| VIH | 11 (3) |
| Otras etiologías | 10 (3) |
| Edad al diagnóstico (años) | 44 [34-56] |
| Tiempo de seguimiento (años) | 5 [2-8] |
| PM6M (m) | 396 [325-475] |
| Parámetros hemodinámicos | |
| Presión arterial pulmonar media (mmHg) | 56 ± 15 |
| Presión auricular derecha (mmHg) | 9 ± 5 |
| RVP (UW) | 13 ± 7 |
| IC (l/min/m2) | 2 ± 0,8 |
| Clase funcional | |
| I-II | 105 (28) |
| III-IV | 274 (72) |
| Ecocardiografía | |
| Derrame pericárdico | 54 (14) |
| Tipo de tratamiento | |
| Monoterapia | 321 (85) |
| Doble terapia | 42 (11) |
| Triple terapia | 6 (2) |
EVOP: enfermedad venooclusiva pulmonar; HAP: hipertensión arterial pulmonar; PM6M: prueba de marcha de 6 minutos;; RVP: resistencia vascular pulmonar; UW: unidades Wood; VIH: virus de la inmunodeficiencia humana.
Salvo otra indicación, los datos expresan n (%), media ± desviación estándar o mediana [intervalo intercuartílico].
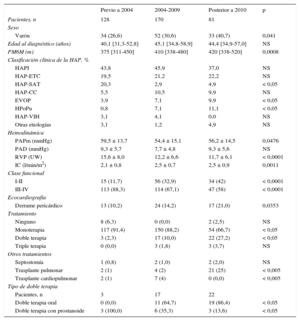
Características basales según el intervalo de tiempo del diagnóstico
| Previo a 2004 | 2004-2009 | Posterior a 2010 | p | |
|---|---|---|---|---|
| Pacientes, n | 128 | 170 | 81 | |
| Sexo | ||||
| Varón | 34 (26,6) | 52 (30,6) | 33 (40,7) | 0,041 |
| Edad al diagnóstico (años) | 40,1 [31,3-52,8] | 45,1 [34,8-58,9] | 44,4 [34,9-57,0] | NS |
| PM6M (m) | 375 [311-450] | 410 [338-480] | 420 [338-520] | 0,0008 |
| Clasificación clínica de la HAP, % | ||||
| HAPI | 43,8 | 45,9 | 37,0 | NS |
| HAP-ETC | 19,5 | 21,2 | 22,2 | NS |
| HAP-SAT | 20,3 | 2,9 | 4,9 | < 0,05 |
| HAP-CC | 5,5 | 10,5 | 9,9 | NS |
| EVOP | 3,9 | 7,1 | 9,9 | < 0,05 |
| HPoPu | 0,8 | 7,1 | 11,1 | < 0,05 |
| HAP-VIH | 3,1 | 4,1 | 0,0 | NS |
| Otras etiologías | 3,1 | 1,2 | 4,9 | NS |
| Hemodinámica | ||||
| PAPm (mmHg) | 59,5 ± 13,7 | 54,4 ± 15,1 | 56,2 ± 14,5 | 0,0476 |
| PAD (mmHg) | 9,3 ± 5,7 | 7,7 ± 4,8 | 9,3 ± 5,6 | NS |
| RVP (UW) | 15,6 ± 8,0 | 12,2 ± 6,6 | 11,7 ± 6,1 | < 0,0001 |
| IC (l/min/m2) | 2,1 ± 0,8 | 2,5 ± 0,7 | 2,5 ± 0,9 | 0,0011 |
| Clase funcional | ||||
| I-II | 15 (11,7) | 56 (32,9) | 34 (42) | < 0,0001 |
| III-IV | 113 (88,3) | 114 (67,1) | 47 (58) | < 0,0001 |
| Ecocardiografía | ||||
| Derrame pericárdico | 13 (10,2) | 24 (14,2) | 17 (21,0) | 0,0353 |
| Tratamiento | ||||
| Ninguno | 8 (6,3) | 0 (0,0) | 2 (2,5) | NS |
| Monoterapia | 117 (91,4) | 150 (88,2) | 54 (66,7) | < 0,05 |
| Doble terapia | 3 (2,3) | 17 (10,0) | 22 (27,2) | < 0,05 |
| Triple terapia | 0 (0,0) | 3 (1,8) | 3 (3,7) | NS |
| Otros tratamientos | ||||
| Septostomía | 1 (0,8) | 2 (1,0) | 2 (2,0) | NS |
| Trasplante pulmonar | 2 (1) | 4 (2) | 21 (25) | < 0,005 |
| Trasplante cardiopulmonar | 2 (1) | 7 (4) | 0 (0,0) | < 0,005 |
| Tipo de doble terapia | ||||
| Pacientes, n | 3 | 17 | 22 | |
| Doble terapia oral | 0 (0,0) | 11 (64,7) | 19 (86,4) | < 0,05 |
| Doble terapia con prostanoide | 3 (100,0) | 6 (35,3) | 3 (13,6) | < 0,05 |
CC: cardiopatía congénita; ETC: enfermedad del tejido conectivo; EVOP: enfermedad venooclusiva pulmonar; HAP: hipertensión arterial pulmonar; HAPI: HAP idiopática; HPoPu: hipertensión portopulmonar; IC: índice cardiaco; NS: no significativo; PAD: presión auricular derecha; PAPm: presión arterial pulmonar media; PM6M: prueba de marcha de 6 minutos;; RVP: resistencias vasculares pulmonares; SAT: síndrome de aceite tóxico; UW: unidades Wood; VIH: virus de la inmunodeficiencia humana.
Salvo otra indicación, los datos expresan n (%), media ± desviación estándar o mediana [intervalo intercuartílico].

Distribución de los pacientes según percentil de edad. CC: cardiopatías congénitas; ETC: enfermedad del tejido conectivo; EVOP: enfermedad venooclusiva pulmonar; HAPH: hipertensión arterial pulmonar heredable; HAPI: hipertensión arterial pulmonar idiopática; HPoPu: hipertensión portopulmonar; SAT: síndrome de aceite tóxico; VIH: virus de la inmunodeficiencia humana.
El tratamiento inicial con prostanoides en monoterapia ha disminuido con el tiempo; desde la introducción de la terapia oral, se ha restringido a pacientes con perfil de alto riesgo. Se trató a los pacientes con prostaciclinas una mediana de 7,9 [3,8-13,2] años antes del deterioro clínico (tabla 2). Se incrementó el tratamiento combinado de inicio (el 2,3, el 10 y el 27,2% en el primero, el segundo y el tercer periodo respectivamente) (p < 0,05).
Se incluyó a 48 pacientes en lista de espera de trasplante y se han realizado 36 trasplantes: 9 cardiopulmonares y 27 bipulmonares (figura 1B). Del total de pacientes incluidos en lista de espera de trasplante, se diagnosticó en el primero, el segundo y el tercer periodo al 22, el 43 y el 35% respectivamente (p < 0,05). Todos los pacientes estaban tratados con prostaciclinas sistémicas antes de su inclusión en lista de espera de trasplante (excepto los pacientes con EVOP). El número y el tipo de trasplante realizado variaron: el 14% (2 cardiopulmonares y 3 bipulmonares), el 17% (5 cardiopulmonares y 1 bipulmonar) y el 69% (todos bipulmonares) de todos los trasplantes en el primero, el segundo y el tercer periodo respectivamente. A partir de 2011, los TP se han realizado en nuestro centro y 1 paciente con disfunción biventricular fue remitido para valoración de trasplante cardiopulmonar a otro hospital y permanecía en lista de espera a fin de seguimiento.
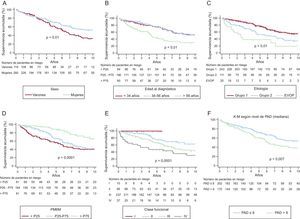
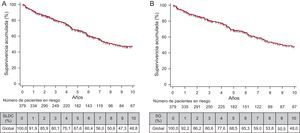
Análisis de supervivencia y factores pronósticosLa mediana de seguimiento fue de 4,5 [2,2-8,2] años y se perdieron 14 casos (4%). La figura 3 muestra variables que en el análisis bivariable presentaron diferencias significativas en deterioro clínico y después se incluyeron en el análisis multivariable. La supervivencia libre de deterioro clínico fue mayor entre las mujeres, a edad < 56 años, con presión auricular derecha < 8mmHg, con PM6M > 475 m y en CF I–II (figura 3). En relación con la etiología de la HAP, el grupo de peor supervivencia fue el 3 (figura 3C). La mediana de supervivencia libre de trasplante o muerte fue de 9 años (IC95%, 7,532-11,959). En la figura 4A se muestran datos de supervivencia libre de deterioro clínico (el 91,9, el 80,1 y el 67,6% el primero, el tercero y el quinto año respectivamente) con una mediana de supervivencia de 8,2 años (IC95%, 7,0-11,6). La figura 4B muestra la supervivencia libre de muerte o trasplante el primero, el tercero y el quinto año, que fueron del 92,2, el 80,6 y el 68,5%. La causa de fallecimiento del 52,4% estuvo relacionada con HAP (el 79% insuficiencia cardiaca y el 21% muerte súbita), el 25,4% no estaba relacionada (el 18% infecciones y el 12% neoplasias) y en el 22,2%, indeterminada. La tabla 3 muestra el análisis multivariable de factores pronósticos de deterioro clínico. El C–index para el modelo multivariable fue 0,8 (IC95%, 0,66-0,91).

. B: supervivencia libre de muerte o trasplante. SG: supervivencia global.")
Análisis multivariable: factores pronósticos de deterioro clínico al momento del diagnóstico
| HR (IC95%) | p | |
|---|---|---|
| Variables demográficas | ||
| Varón | 1,52 (1,09-2,12) | 0,0142 |
| Edad > 56 años | 1,95 (1,38-2,77) | 0,0002 |
| Etiología (ómnibus) | 0,0003a | |
| Etiología del grupo 2 (HAP-ETC, HPoPu, otras) | 1,51b (1,06-2,14) | 0,0221 |
| Etiología del grupo 3 (EVOP) | 2,87b (1,67-4,92) | 0,0001 |
| Criterios compuestos | ||
| Criterio 1c | 1,46 (1,03-2,07) | 0,0334 |
| Criterio 2d | 1,59 (1,15-2,20) | 0,0047 |
CF: clase funcional; ETC: enfermedad del tejido conectivo; EVOP: enfermedad venooclusiva pulmonar; HAP: hipertensión arterial pulmonar; HR: hazard ratio; HPoPu: hipertensión portopulmonar; IC95%: intervalo de confianza del 95%; PAD: presión auricular derecha; PM6M: prueba de marcha de 6 minutos; VIH: virus de la inmunodeficiencia humana.
Nuestro estudio es la primera serie publicada que refleja el cambio en el tratamiento de HAP durante un periodo de 30 años e incluye una cohorte consecutiva de pacientes con HAP atendidos en un centro de referencia nacional con alto volumen de pacientes y aporta información sobre la supervivencia > 5 años. En estas 3 décadas, se observa una modificación en las etiologías de la HAP, acumulación de HAP-aceite de colza antes de 2004, y destaca el diagnóstico de nuevos casos en el último periodo, tras un largo tiempo desde la exposición. Se observa una concentración de pacientes más complejos después de 2010, y los aumentos de EVOP y de hipertensión portopulmonar son los cambios más destacados. Este hecho se relaciona con la adaptación de la estructura de la unidad a nuevas exigencias, con la incorporación de: cribado de hipertensión portopulmonar de los pacientes candidatos a trasplante hepático, cribado de HAP-esclerodermia, estudio genético de nuestra población y el inicio del programa de TP a partir de 2011. El trabajo multidisciplinario en el cribado de poblaciones de pacientes con enfermedades asociadas a HAP y familiares de pacientes con formas heredables de la enfermedad ha permitido mejorar en el diagnóstico precoz. A partir de 2004, se observa menor gravedad hemodinámica y mejor CF en el momento del diagnóstico. Por este motivo, y sumado a la experiencia de otros grupos7,8, el diagnóstico precoz y la intervención temprana en la HAP pueden traducirse en mejores resultados a largo plazo.
Hipertensión arterial pulmonar idiopáticaEs la etiología más frecuente. La mediana de edad al diagnóstico de 44 años ha permanecido sin variaciones en el tiempo. En contraposición, los registros COMPERA8 (2007-2012) y de Reino Unido9 (2001-2009) mostraron una mediana al diagnóstico de 71 y 62 años respectivamente. Este hecho podría explicarse por la falta de derivación de pacientes añosos a nuestro centro en los últimos años, en relación con el creciente número de unidades de HAP en España.
La mediana de supervivencia libre de muerte o trasplante fue de 11 años, superior a los 7 años comunicados por el registro REVEAL6. Se ha descrito que la edad avanzada, CF III o IV y la distancia recorrida en la PM6M son factores pronósticos establecidos en pacientes con HAP idiopática6. En nuestra serie, los pacientes son más jóvenes, tienen mejor CF, recorren más distancia en la PM6M y se utilizan más prostaciclinas, lo que puede contribuir a las diferencias observadas en la supervivencia.
Hipertensión arterial pulmonar-enfermedad del tejido conectivoEl 50% de los pacientes mayores de 56 años tiene HAP-enfermedades del tejido conectivo y el 92% de ellos, esclerodermia. En el registro REVEAL10, al igual que en nuestra serie, los pacientes con esclerodermia tienen una media de edad de 62 años y constituyen el grupo más prevalente entre los pacientes mayores de 50 años. La supervivencia al primer año de pacientes con HAP-esclerodermia en nuestra serie es del 81%, superponible a la descrita en el registro REVEAL10. La mortalidad en el primer año está próxima al 20% y es una llamada de atención a la comunidad médica que ha llevado a proponer recientemente nuevos protocolos de cribado11 y tratamiento, que se han implementado en nuestra unidad en el último año. Conseguir un diagnóstico precoz es la clave para mejorar la supervivencia12.
Hipertensión arterial pulmonar asociada a aceite de colzaLa epidemia relacionada con la ingesta de aceite de colza desnaturalizado ocurrió en 1981 y afectó a cerca de 20.000 personas. Se desarrolló HAP en aproximadamente el 20% de afectados, con remisión espontánea aparentemente total en muchos de ellos, y en un pequeño porcentaje evolucionó a una forma crónica y grave1. Nuestro hospital centralizó el seguimiento de pacientes con este tipo de HAP, que se diagnosticó en el 74% de los casos antes de 2004. Sin embargo, se producen nuevos diagnósticos, algunos de ellos con intervalos entre la exposición y la aparición de HAP > 30 años. Estos periodos de latencia de duración variable también se describen en el desarrollo de HAP tras la ingesta de anorexígenos. Así, en la serie del registro francés, se diagnosticó a un 44% de los pacientes13 tras más de 5 años desde la exposición al fármaco. Por lo tanto, el antecedente de ingesta de aceite de colza debe estar presente en nuestro medio y ser identificado como factor de riesgo propio en España, a pesar del largo tiempo de latencia.
Hipertensión arterial pulmonar-cardiopatías congénitasSe excluyó a los pacientes en situación de Eisenmenger por tener una historia natural y un tratamiento terapéutico diferentes. Más de la mitad del resto de pacientes presentaban formas asociadas a reparaciones de shunt sistémico-pulmonares realizadas en la infancia, y el diagnóstico de HAP se había establecido en la cuarta década de la vida. En el REHAP14 se describe que un 25% de los 240 pacientes con HAP-cardiopatía congénita presentaron hipertensión pulmonar tras la reparación y, al igual que en nuestra serie, la supervivencia fue superponible a la de la HAP idiopática.
El segundo grupo clínico más frecuente es el asociado a defectos restrictivos (comunicación interauricular en su mayoría), con una supervivencia superponible a la de la HAP idiopática. En pacientes que recibieron TP por HAP-cardiopatía congénita simple con shunt sistémico-pulmonar, se procedió al cierre del defecto durante el TP. Nuestra unidad es pionera en el país en la reparación de la cardiopatía congénita en el momento del trasplante, tal y como recomiendan las actuales guías de trasplante15.
Enfermedad venooclusiva pulmonarLa EVOP se caracteriza por una progresión rápida y carecer de opciones de tratamiento. Mutaciones en el gen EIF2AK4 se han relacionado con el desarrollo de esta enfermedad, con un patrón de herencia autosómico recesivo. Recientemente nuestro grupo ha descubierto una mutación fundadora en dicho gen EIF2AK4 (c.3344C>T [p.P1115L]) en 5 familias de etnia gitana con EVOP caracterizada por aparición de la enfermedad a una edad < 35 años, consanguinidad, baja capacidad de difusión de monóxido de carbono y corta supervivencia16.
En nuestra serie, el 75% de los pacientes con EVOP llegaron a nuestro centro en estado crítico y el 50% de ellos requirieron apoyo con oxigenador extracorpóreo de membrana en los primeros 3 meses desde el diagnóstico, sin fallecimientos en lista de espera. En este aspecto, otros grupos han descrito una alta tasa de mortalidad de estos pacientes en lista de espera, hasta el 22% a los 6 meses17. El corto tiempo de evolución desde el diagnóstico y la ausencia de tratamiento específico hacen imprescindible la derivación inmediata ante la sospecha de EVOP, para iniciar la valoración por el equipo de TP. En caso de formas hereditarias, se debe hacer el estudio genético y cribado a familiares de primer grado3 e identificar correctamente a los portadores heterocigotos. Por lo tanto, este hallazgo obliga a estar especialmente alerta en España y no demorar el diagnóstico ante una clínica sospechosa en pacientes de este grupo étnico.
ProstaciclinasSe utilizaron prostaciclinas en 205 pacientes (56%). El 70% de los que fallecieron y el 75% de los pacientes en CF IV se encontraban en tratamiento con prostaciclinas. En el registro REVEAL, la utilización de prostaciclinas se redujo drásticamente y solo el 43% de los fallecidos estaban tratados18. En el REHAP, desde una perspectiva nacional, se observó una infrautilización, de modo que únicamente el 50% de los pacientes en CF IV recibieron prostaciclinas2. Esta diferencia entre datos de registros poblacionales y un centro experto se explica por la necesidad de una organización asistencial compleja que dé respaldo a la utilización de estos fármacos y garantice la seguridad y la excelencia en su tratamiento. Es imprescindible enfermería entrenada, un programa de educación sanitaria estructurado y un sistema de respuesta inmediata (presencial y telefónica) las 24 h.
TrasplanteLa inclusión en lista de TP depende de múltiples factores, fundamentalmente la velocidad de progresión de la enfermedad y la escasez de órganos que conlleva una duración cada vez mayor en la lista de espera. Por ello, en las últimas guías clínicas3 se incluyeron 2 elementos diferenciados: a) la derivación para valoración de TP debe ser precoz antes de agotar todas las posibilidades terapéuticas, y b) la inclusión activa en el programa de TP. Se recomienda desarrollar estrategias de puente al trasplante para minimizar la mortalidad en lista de espera. En nuestra unidad, a partir de 2011 se realizó en código urgente un 15% de los TP por HAP y en la mitad se ha necesitado una estrategia puente al TP, fundamentalmente el soporte con oxigenador extracorpóreo de membrana en 4 pacientes. La septostomía auricular ha permitido la estabilización clínica del paciente y la realización electiva del TP con éxito en 5 pacientes19.
Factores predictores de riesgoLas variables demográficas no modificables y con impacto pronóstico subrayan la necesidad de establecer estrategias terapéuticas desde el inicio para pacientes con un perfil de alto riesgo. En nuestra serie, las variables pronósticas modificables relacionadas con la evolución de la enfermedad, como la CF III-IV, la PM6M < 475 m y una presión de la aurícula derecha > 8mmHg (incluidos en los criterios compuestos), eran superponibles a los de la mayoría de los registros1,6 y respaldan la recomendación de las guías clínicas de realizar valoraciones por objetivos5.
Asimismo se ha realizado un análisis con variables compuestas que discrimina a los pacientes en CF III-IV de aquellos que tienen mayor riesgo y podrían necesitar un tratamiento más agresivo. Kane et al.20 exploraron esta estrategia en una cohorte de 484 pacientes; la estimación pronóstica multifactorial conseguía un modelo con buena capacidad de predicción, con un C-index de 0,84. Nuestro modelo multivariable, con la incorporación de criterios compuestos (CF + PM6M y CF + presión auricular derecha) consigue un C-index de 0,80, lo que resalta la importancia de la valoración multifactorial en la estimación del pronóstico, tal y como se recomienda en las guías de práctica clínica actuales5.
LimitacionesEs un estudio observacional ambispectivo (se incluyó retrospectivamente en el estudio al 12% de los pacientes) con la experiencia de un centro que incluye una cohorte de pacientes durante un largo intervalo de tiempo, con marcadas diferencias en las etiologías de la HAP y las opciones terapéuticas disponibles en cada periodo. El análisis de los factores de riesgo y la supervivencia debe interpretarse con precaución, ya que existe un sesgo temporal. Sin embargo, creemos que una descripción de la realidad clínica observada en un centro de referencia durante 3 décadas permite una visión global de la enfermedad en el tiempo, y puede ayudar a establecer estrategias dirigidas a mejorar la atención de esta enfermedad. Al ser una serie de 30 años, no se han utilizado las nuevas herramientas para la estratificación pronóstica incorporadas a partir de 2010: fracción aminoterminal del propéptido natriurético cerebral, parámetros ergoespirométricos y las nuevas técnicas de imagen.
CONCLUSIONESNuestra serie presenta la mayor cohorte de pacientes con HAP y el mayor tiempo de seguimiento en España descritos hasta la fecha. El tratamiento de la enfermedad ha sufrido una drástica evolución que ha permitido el diagnóstico precoz en poblaciones de riesgo y el tratamiento de alta complejidad, que incluye terapia farmacológica combinada, asistencias como el oxigenador extracorpóreo de membrana y el TP. El presente estudio amplía la información disponible de patrones de supervivencia a largo plazo y proporciona nueva información sobre variables clínicas compuestas y la utilidad de la integración de estas variables en la estimación del riesgo de deterioro clínico.
FINANCIACIÓNBeca educativa no condicionada por ACTELION. Los fondos para este estudio fueron aportados por el Instituto de Salud Carlos III y el Ministerio de Economía y Competitividad a través de CIBER en enfermedades cardiovasculares (CB16/11/00502).
CONFLICTO DE INTERESESP. Escribano Subías ha recibido una beca educacional no condicionada de Actelion para el estudio estadístico realizado Xail (biometría para análisis estadístico); ha sido consultora de Actelion, Bayer, Pfizer y GSK; ha recibido becas de Actelion y GSK; remuneración por desarrollo de ponencias por parte de Actelion, Bayer, Pfizer y GSK, y remuneración por desarrollo de presentaciones educativas por parte de GSK. P.E. Carreira ha recibido una beca de ISCIII-FIS y remuneración por desarrollo de ponencias por parte de Actelion. J.F. Delgado Jiménez ha recibido remuneración por desarrollo de ponencias por parte de Actelion y Bayer. M.Á. Gómez Sánchez ha sido consultor para Actelion, MSD, Ferrer, GSK y Bayer, y ha recibido remuneración por desarrollo de ponencias y de presentaciones educativas de Actelion, MSD, Ferrer, GSK y Bayer. El resto de los autores declara no tener ningún conflicto de intereses.
- –
Los datos epidemiológicos aportados por los grandes registros de HAP han permitido discernir factores pronósticos. Las actuales guías de práctica clínica recomiendan una valoración periódica y multifactorial de la enfermedad. La supervivencia, según datos del registro español de HAP, es del 65% a 5 años.
- –
Nuestro estudio refleja la evolución en la epidemiología y el tratamiento de la HAP durante un periodo de 30 años que incluye una cohorte consecutiva de pacientes con HAP atendidos en un centro de referencia nacional con alto volumen de pacientes. El análisis multivariable y las variables compuestas confirman la necesidad de un seguimiento apoyado en la valoración multifactorial periódica de la enfermedad. Aporta información sobre supervivencia > 5 años en el seguimiento.
A Xavier Masramón, de Xail Biometría.