La miocardiopatía arritmogénica predominantemente izquierda (MCAI) presenta características fenotípicas y genotípicas reflejadas en la familia española de cinco miembros que aquí describimos. Durante un catarro, un joven presentó una taquicardia ventricular con origen ventricular izquierdo y realce tardío en dicha localización. Su ECG basal mostró bajos voltajes, retraso en la activación terminal del QRS (cara inferior y V4-V6) y trastorno de la conducción auriculoventricular. Su biopsia endomiocárdica evidenció pérdida miocitaria y fibrosis. Aunque inicialmente fue catalogado de miocarditis, la evaluación familiar fue decisiva para sospechar una MCAI. El estudio genético identificó una mutación nueva en desmoplaquina tipo «sin sentido» (Q1866X) congruente con la presencia de una desmoplaquina truncada en muestras de piel de los afectados.
Palabras clave
La miocardiopatía arritmogénica (MCA) tiene una prevalencia de 1:5.0001. Un 40% de los casos presenta mutaciones, la mayoría en los genes que codifican para las proteínas desmosómicas, habitualmente con herencia autosómica dominante1, 2 y una penetrancia variable. El restante 60% se estima asociado a genes aún no identificados o a causas adquiridas3.
Los desmosomas garantizan la adhesión celular y abundan en tejidos sujetos a tensión mecánica constante, como la piel y el miocardio. La participación de proteínas anómalas en los desmosomas de pacientes con MCA disminuye su adhesividad y favorece la pérdida miocitaria, la sustitución fibroadiposa y la inflamación. Estos hallazgos confieren el sustrato anatómico para la generación de taquiarritmias ventriculares y muerte súbita3.
Originalmente descrita en el ventrículo derecho (VD), la MCA ha ampliado su espectro también a formas que afectan fundamentalmente al ventrículo izquierdo (VI), llamadas miocardiopatía arritmogénica izquierda (MCAI)4, 5, 6.
Presentamos a una familia española estudiada tras un episodio de taquicardia ventricular monomorfa sostenida (TVMS) en el probando. El protocolo cardiológico y genético diagnosticó de MCAI al probando y a un familiar, de portadores genéticos a otros dos y de sujeto sano a otro. Se destacan los rasgos característicos de esta miocardiopatía con el objetivo de facilitar su reconocimiento y enriquecer bases de datos con correlaciones genotipo-fenotipo.
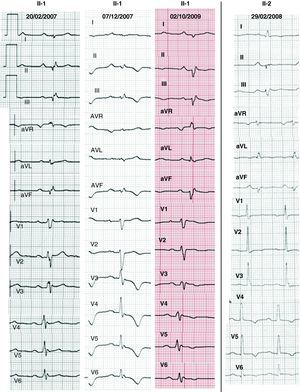
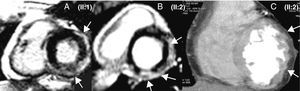
MétodosPacientesEl probando de 36 años (Figura 1, II:1) consultó por dolor torácico atípico por vía ambulatoria; el test de esfuerzo (TE) con protocolo de Bruce fue negativo y su cardiorresonancia magnética (CRM) se informó como miocarditis por presentar realce tardío de gadolinio (RTG) en VI. Ingresó 1 año después durante un catarro por disnea, síncope y una TVMS mal tolerada con morfología de bloqueo de rama derecha y eje superior, que fue cardiovertida. El ECG mostró bajos voltajes, retraso en la activación terminal del QRS de cara inferior y V4-V6 (ocasionalmente también V1-V3) y trastorno de la conducción auriculoventricular (Figura 2). La CRM fue superponible a la previa, con ligera disfunción sistólica del VI y el margen ondulado en la tomografía computarizada de 64 multidetectores (TCMD) indicó infiltración miocárdica por grasa/fibrosis epicárdica (Figura 3). Se realizó una biopsia endomiocárdica (BEM), que mostró fibrosis intersticial y pérdida miocitaria < 30% sin grasa patológica. Se le implantó un desfibrilador y se le dio el alta.

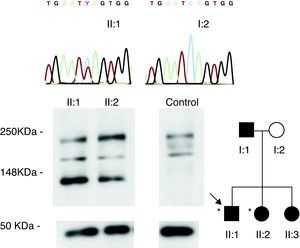
Figura 1. Familiares estudiados (círculos: mujeres; cuadrados: varones; *fenotipo de miocardiopatía arritmogénica izquierda). Se muestran los cromatogramas de los portadores de la mutación desmoplaquina Q1866X (símbolos negros) y de los sujetos sanos (símbolos blancos), así como su patrón de expresión de desmoplaquina en piel.

Figura 2. Electrocardiograma de los pacientes afectados, que muestra bajos voltajes, trastorno de la conducción auriculoventricular y retraso en la activación terminal del QRS de cara inferior y V4-V6 (ocasionalmente también en V1-V3). Las alteraciones de la repolarización (especialmente V4-V6) fueron intermitentes en el probando.

Figura 3. A y B: cardiorresonancia magnética. C: tomografía computarizada multidetectores. Las flechas señalan el realce tardío de gadolinio (A y B) e indican infiltración miocárdica por fibrosis/grasa epicárdica (C).
Aunque con este estudio clínico el probando no cumplía criterios de MCA7, la sospecha de una MCAI motivó una evaluación familiar (padres y dos hermanas) con ECG, ecocardiografía, TE, Holter ECG de 24 h, CRM y bioquímica general.
Completada la valoración familiar, efectuamos un cribado mutacional en el probando, con estudio genético familiar en cascada, tras el cual se realizó una biopsia de piel en los pacientes con MCAI (II:1 y II:2) para estudiar su patrón de expresión de desmoplaquina en comparación con el de un sujeto control.
Estudio genéticoSe extrajo el ADN de leucocitos de sangre periférica. En el probando se secuenciaron en doble sentido los cinco genes desmosómicos principales (placoglobina, placofilina-2, desmogleína-2, desmocolina-2 y desmoplaquina) (ABI Prism 3100 sequencer, Applied Biosystems). Identificada la mutación, en los familiares y 200 cromosomas controles se secuenció exclusivamente el exón afectado.
Estudio tisularLa expresión de desmoplaquina en los afectados y un control sano se analizó mediante inmunotransferencia en biopsias de piel procesadas, como se ha descrito previamente6, con gel Novex 4-12% Tris-Glycine gel 1 mm (Invitrogen) y con la gliceraldehído 3-fosfato deshidrogenasa (GAPDH) como control de carga. Los anticuerpos primarios fueron NW161 (desmoplakin N-terminal specific antibody, donado por la Dra. Kathleen Green, Northwestern University, Evanston, Illinois, Estados Unidos) y GAPDH (14C10) de conejo mAb (IZASA), y los secundarios IgG de oveja antirratón HRP (Amersham Bioscience) e IgG de conejo antiburro HRP (Amersham Bioscience).
El protocolo fue previamente autorizado por el comité ético de nuestro hospital y cada sujeto firmó su consentimiento informado.
ResultadosProbandoEl estudio genético identificó una variante nueva en el gen de la desmoplaquina (c. 5596C>T, Q1866X) (Figura 1), ausente en controles, que originaba un codón de parada en la traducción del dominio rod central de la proteína. La inmunotransferencia detectó una desmoplaquina truncada en su biopsia de piel (Figura 1).
Estudio familiarNo existían antecedentes familiares destacables. El protocolo cardiológico en sus padres y una hermana fue normal (Tabla 1). Sin embargo, su otra hermana presentó fenotipo de MCAI (II:2) con ECG, CRM y TCMD similares a los del probando (Figura 2, Figura 3). Se realizó un estudio electrofisiológico que documentó la presencia de potenciales fragmentados en VD y una TVMS (con bloqueo de rama izquierda y eje inferior) fácilmente inducible. Su patrón de expresión de desmoplaquina en piel fue superponible al del probando (Figura 1). Tras estos resultados, se le implantó un desfibrilador. La mutación en desmoplaquina Q1866X, ausente en la madre del probando (I:2), estaba presente en la hermana con el fenotipo de MCAI (II:2) y en 2 portadores genéticos con estudio cardiológico normal (la otra hermana II:3 y el padre I:1). Con los resultados genéticos, los dos pacientes afectados (II:1 y II:2) cumplían los nuevos criterios de MCA8.
Tabla 1. Resultados del estudio familiar
| Paciente | Sexo/edad (años) | Historia | ECG | Ecocardiograma | Analítica | TE | Holter ECG de 24 h | CRM | TCMD | BEM | EEF | Genética | Expresión de DSP |
| I:1 | V/77 | - | RS, BAV 1°, hemibloqueo anterior | Normal | Normal | No submáxima negativa | RS, 0 EVs | Normal | _ | _ | _ | DSP Q1866X en heterocigosis | _ |
| I:2 | M/75 | Disnea II/IV NYHA | RS, HVI | IA ligera | Normal | Submáxima negativa | RS, 0 EVs | Normal | _ | _ | _ | Sin mutación | - |
| II:1 (probando) | V/37 | Dolor torácico atípico. DAI | RS, bajos voltajes, BAV 1°, retraso de la activación terminal del QRS en cara inferior y V1-6, T negativo en V3-6 | Normal | (transitorio de CK [128 U/l]), CK-MB masa (5 U/l) y transminasas (AST 129 U/l, ALT 176 U/l y LDH 758 U/l) durante el ingreso por TVS | Submáxima negativa, 7 EVs (BRD y BRI) | _ | Grosor y volúmenes normales; FEVI, 54% y FEVD, 42%. RTG subepicárdico en cara inferior, lateral y anterior del VI, signo del acordeón en el VD. Sin infiltración grasa | _ | Fibrosis intersticial con pérdida miocitaria < 30% | _ | DSP Q1866X en heterocigosis | DSP I, DSP II y banda de DSP truncada a 160 kDa |
| II:2 | M/41 | Palpitationes, síncopes vasovagales, síndrome de Sjögren, enfermedad de Raynaud y probable lupus eritematoso sistémico. DAI | RS, PR límite, bajos voltajes, transición precordial en V1, retraso de la activación terminal del QRS en cara inferior, T aplanadas en V1-3 y negativas en V4-6 | HK global, FEVI 45% | Normal | No submáxima negativa, BAV 1°, 4 EVs (BRD y BRI) | RS, 636 EVs | HK global, adelgazamiento parcheado del VI; FEVI 42%; IVTSVI 54 ml/m2; FEVD 52%. RTG septal en VD y subepicárdico en VI inferoseptal, inferior y lateral. Sin infiltración grasa | Miocardio del VI ondulado, que indica infiltración fibrosa/grasa | _ | AH 110, HV 41. Potenciales fragmentados de VD, TVMS inducible (BRI con eje inferior) | DSP Q1866X en heterocigosis | DSP I, DSP II y banda de DSP truncada a 160 kDa |
| II:3 | M/40 | Síncopes vasovagales | RS | Normal | Normal | Submáxima negativa | RS, 64 EVs | Normal | _ | _ | _ | DSP Q1866X en heterocigosis | _ |
BAV: bloqueo auriculoventricular; BEM: biopsia endomiocárdica; BRD: bloqueo de rama derecha; BRI: bloqueo de rama izquierda; CRM: cardiorresonancia magnética; DAI: desfibrilador automático implantable; DSP: desmoplaquina; ECG: electrocardiograma; EEF: estudio electrofisiológico; EV: extrasistolia ventricular; FE: fracción de eyección; HK: hipocinesia; HVI: hipertrofia ventricular izquierda; IA: insuficiencia aórtica; IVTSVI: volumen telesistólico ventricular izquierdo indexado; M: mujer; NYHA: New York Heart Association; RS: ritmo sinusal; RTG: realce tardío de gadolinio; TCMD: tomografía computarizada multidetectores; TE: test de esfuerzo; TVMS: taquicardia ventricular monomorfa sostenida; V: varón; VD: ventrículo derecho; VI: ventrículo izquierdo.
Este trabajo destaca la importancia del estudio multidisciplinario al evaluar a familias con sospecha de MCA y describe una mutación nueva en el gen de la desmoplaquina apuntando al mecanismo molecular más probablemente implicado.
El ECG de la MCA con afectación del VI presenta aplanamientos/inversiones de la repolarización (infero-)laterales4, 5, 6, 8. Además, nuestros pacientes presentaban bajos voltajes y un retraso tanto en la activación ventricular de cara inferior como en la conducción auriculoventricular. Estos novedosos hallazgos, no recogidos en otros artículos4, 5, 6 ni en los nuevos criterios8, fueron destacados en un caso con enfermedad de Carvajal9 y podrían ser útiles para sospechar una MCAI. La localización electrocardiográfica de estas alteraciones coincide con la extensa fibrosis detectada en la cara inferolateral.
De acuerdo con las primeras impresiones10, la inclusión del diagnóstico genético aumentó la sensibilidad de los criterios de MCA aunque, desgraciadamente, su accesibilidad para el cardiólogo clínico es todavía limitada. Si bien se han descrito ocasionalmente mutaciones en desmocolina-2 y desmogleína-2, nuestros resultados refuerzan que el de la desmoplaquina podría ser el primer gen que estudiar ante una MCAI4, 11. La inmunotransferencia comprobó la predicción del truncamiento de la desmoplaquina en presencia de la mutación Q1866X. Así, la ausencia de región C-terminal en el 50% de la desmoplaquina de los afectados justificaría una unión defectuosa entre los desmosomas y los filamentos intermedios4.
Aunque la indicación de un desfibrilador como prevención primaria en la hermana afectada (II:2) era controvertida, le aconsejamos este tratamiento por la presencia de marcadores de riesgo como la historia familiar, la afectación del VI y la inducción de TVMS en el estudio electrofisiológico12.
En la MCAI predomina el componente fibroso sobre la infiltración grasa4, 5, 6, con un patrón subepicárdico de RTG similar al de distintas afecciones (miocarditis, miocardiopatías infiltrativas, sarcoidosis, enfermedad de Chagas, miocardiopatía dilatada, distrofinopatías, etc.)13. En nuestro probando, la primera CRM suscitó el diagnóstico de miocarditis, y sin el estudio familiar no se habría filiado correctamente. Debe recordarse que las miocarditis pueden actuar como desencadenantes de los brotes de actividad de la MCA y que la ausencia de datos histológicos típicos de MCA no descarta el diagnóstico. El patrón ondulante que observamos en la TCMD corrobora los hallazgos de la CRM y podría ser útil en los pacientes portadores de un desfibrilador.
En resumen, el diagnóstico de MCAI es complejo, con exploraciones no siempre accesibles, y la evaluación familiar puede ser una pieza clave en el diagnóstico diferencial.
FinanciaciónEste trabajo se ha financiado con ayudas del Instituto de Salud Carlos III (PI070831, CP0700326, RECAVA RD06/0014/0004), la Sociedad Valenciana de Cardiología, la Heart Rhythm Society y la British Heart Foundation.
Conflicto de interesesNinguno.
Agradecimientos
Agradecemos la colaboración de los Dres. Monserrat Évole, Aitana Braza-Boïls y Antonio Moscardó en la ejecución de las técnicas utilizadas.
Recibido 27 Abril 2010
Aceptado 12 Octubre 2010
Autor para correspondencia: Departamento de Cardiología, Hospital La Fe, Avda. Campanar 21, 46009 Valencia, España. zorio_est@gva.es