Palabras clave
INTRODUCCION
La miocardiopatía hipertrófica (MCH) es una cardiopatía de transmisión genética caracterizada por el engrosamiento de la pared miocárdica en ausencia de otras causas de hipertrofia ventricular izquierda1. Es una de las causas más frecuentes de muerte súbita en jóvenes y la más común de las enfermedades hereditarias cardiovasculares, con una prevalencia estimada de una de cada 500 personas2.
Hasta el momento se han identificado más de 400 mutaciones diferentes causantes de MCH en 11 genes que codifican proteínas del sarcómero cardiaco3. Uno de estos genes es MYBPC3, que codifica la proteína C fijadora de miosina. Tradicionalmente se ha considerado que las mutaciones en el gen MYBPC3 causan una MCH de presentación más tardía, con menor hipertrofia ventricular y mejor pronóstico que las causadas por mutaciones en otros genes sarcoméricos4-6.
Describimos el caso de una familia con varios miembros afectados de MCH y alta incidencia de muerte súbita, que presenta una mutación en el gen MYBPC3 no descrita con anterioridad7.
MÉTODOS
Estudiamos a los sujetos vivos de una rama de una familia con múltiples antecedentes de muerte súbita y cardiopatía (fig. 1). El estudio incluyó exploración física, electrocardiograma y ecocardiograma. A los miembros con MCH se les practicaron, además, una ergometría y un registro electrocardiográfico de 48 horas.

Fig. 1. Árbol genealógico familiar. Círculo: mujer. Cuadrado: varón. Símbolos rojos: mi- embros de la familia afectados de cardiopatía/muerte súbita. Símbolos marrones: miembros sin cardiopatía. Símbolos que encierran un círculo: miembros portadores de la mutación sin cardiopatía en la actualidad. Símbolos con línea diagonal: miembros fallecidos. Flecha: paciente índice.
Tras establecer el diagnóstico de MCH en la paciente índice, la mayor de sus hijas, de 34 años, fue estudiada en Noruega, donde residía, y fue diagnosticada también de MCH no obstructiva. Tras otorgar el consentimiento escrito, en esta enferma se realizó un estudio genético mediante secuenciación de los exones de los genes de la cadena pesada de la miosina β (MYH7), proteína C fijadora de la miosina (MYBPC3), troponina I (TNNI3), troponina T (TNNT2), cadena ligera de la miosina reguladora 2 (MYL2) y cadena ligera de la miosina esencial 1 (MYL3). Los genes estudiados son causantes de casi todos los casos de MCH con mutación demostrada8.
Tras identificar una mutación en el gen MYBPC3, en la paciente índice y los familiares accesibles de primer grado de los sujetos con MCH o fallecidos de muerte súbita (fig. 1; sujetos II-1, III-1, III-2, IV-2, IV-3, IV-4, IV-7, IV-8 y IV-9), se realizó un estudio genético. Éste consistió en la amplificación mediante la reacción en cadena de la ADN polimerasa (PCR) del exón 2 y las regiones intrónicas adyacentes del gen MYBPC3.
Los cebadores y métodos empleados se encuentran a disposición de quien lo solicite en la dirección de correspondencia. El estudio genético fue aprobado por el Comité de Ética del Hospital La Paz.
RESULTADOS
Evaluación clínica
Sujeto índice
Mujer de 53 años de edad con historia de hipertensión arterial de larga evolución y tabaquismo que es remitida a nuestro centro por disnea progresiva con el esfuerzo. El estudio inicial mostró cardiomegalia radiológica y signos electrocardiográficos de hipertrofia ventricular izquierda. Un ecocardiograma objetivó hipertrofia severa de las paredes anterior, anteroseptal y septal con un grosor del tabique interventricular de 25 mm, contractilidad normal, velocidad de la onda E de 0,71 ms, tiempo de deceleración de la onda E de 130 ms y ausencia de gradiente subaórtico, hallazgos compatibles con el diagnóstico de MCH no obstructiva con disfunción diastólica avanzada. Tras documentarse taquicardias ventriculares no sostenidas (TVNS) en 2 registros electrocardiográficos de 48 h, y dados los antecedentes familiares de muerte súbita, se procedió a la implantación de un desfibrilador automático implantable (DAI).
Historia familiar
El abuelo paterno de la paciente índice, su padre y uno de los tíos paternos habían fallecido súbitamente cerca de los 50 años (52, 49 y 48 años, respectivamente). Dos hermanos varones habían sido diagnosticados de cardiopatía. El primero de ellos falleció súbitamente a los 32 años sin descendencia y el segundo, que era el único con diagnóstico establecido de MCH, falleció a los 42 años tras presentar una hemorragia cerebral cuando se encontraba en espera de trasplante cardiaco. La hija ya comentada de la paciente índice, diagnosticada de MCH a los 34 años, mostró TVNS en el registro electrocardiográfico ambulatorio, por lo que se procedió a la implantación de un DAI. Los otros miembros de la familia estudiados permanecían asintomáticos y presentaban ecocardiogramas normales (fig. 1).
Análisis genético
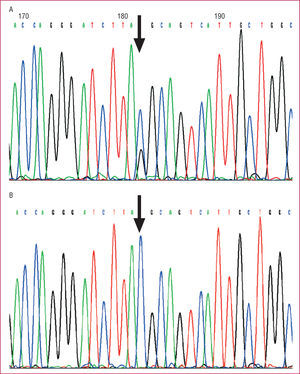
Se constató que la paciente índice y su hija afectada de MCH eran heterocigotas para la mutación Y79X en el exón 2 del gen MYBPC3. En el nucleótido 269 del ARNm de uno de los alelos del gen MYBPC3 el cambio de una base de citosina por guanina (fig. 2) (TAC > TAG) produce un cambio en el codón 79, de modo que en lugar de codificar para el aminoácido tirosina (TAC) se transforma en un codón de terminación (TAG). Esta mutación provoca invariablemente la interrupción de la traducción del ARNm en este punto, dando lugar a la síntesis de una proteína incompleta.

Fig. 2. Secuenciación del exón 2 del gen MYBPC3 de A: paciente índice (flecha en figura 1) y B: hija de la paciente índice no portadora (*en figura 1). A: Se aprecian 2 picos que representan tanto citosina (azul) como guanina (negro) en el punto correspondiente al nucleótido 269 del ARNm del gen MYBPC3. Este doble pico indica que en uno de los alelos el codón TAC, que codifica la tirosina 79 de la proteína, se transforma en TAG (codón de terminación o sin sentido). B: El mismo punto del gen MYBPC3 registra un único pico (citosina, azul) que se corresponde con 2 alelos normales para el codón TAC de la tirosina 79.
Otros 3 miembros de la familia sin cardiopatía en la evaluación clínica (sujetos IV-3, IV-4 y IV-8, de 19, 15 y 28 años, respectivamente) presentaban la misma mutación en el exón 2 de MYBPC3. Los sujetos II-1, III-2, IV-2, IV-7 y IV-9 no presentaban dicha mutación (fig. 1).
DISCUSION
Desde la primera identificación en 1990 de uno de los genes relacionados con la MCH9, el progresivo conocimiento de las bases genéticas de la enfermedad ha revolucionado la atención de estos enfermos y sus familias.
El estudio genético practicado a esta familia ha permitido identificar una nueva mutación en el gen MYBPC3 que se asocia con un curso desfavorable, con desarrollo de la enfermedad y muerte súbita entre la cuarta y sexta décadas de la vida.
Pese a la supuesta benignidad de la MCH secundaria a mutaciones en MYBPC34-6, la presentación en esta familia apoya los hallazgos de otros trabajos, que ligan el curso de la enfermedad al tipo de mutación documentada en este gen10. Así, se ha señalado que los pacientes con mutaciones de terminación en MYBPC3 (como la descrita en este trabajo) presentan la enfermedad más precozmente y requieren terapias más agresivas (ablación septal e implantación de desfibrilador) que los pacientes con mutaciones que provocan solamente cambios en la secuencia de aminoácidos10. En esta familia se implantó un DAI en la paciente índice y en su hija diagnosticada de MCH.
Aunque es probable que la mutación descrita sea la causante de la MCH en esta familia, no se puede asegurar categóricamente, ya que esta mutación no se ha encontrado en otras familias con MCH, y no se han analizado todos los genes causantes de MCH. La influencia de alteraciones en otros genes podría desempeñar un papel fundamental en las manifestaciones clínicas de la enfermedad y podría justificar, en parte, las diferencias observadas en el curso clínico entre los miembros de la familia estudiada. Así, llama la atención que todos los miembros fallecidos eran varones y que fallecieron a una edad más temprana que la del diagnóstico de la paciente índice.
La identificación de la mutación causante en los familiares asintomáticos permite identificar con claridad a los sujetos con posibilidad de desarrollar MCH en el futuro y los que, por no ser portadores de la mutación, no precisan de seguimiento específico11. En esta familia, la hermana de la paciente índice, una hija, 2 sobrinos y los descendientes de todos ellos no precisan de seguimiento en el futuro (fig. 1).
Para los pacientes con MCH, y para los portadores asintomáticos de una mutación, la identificación de la misma posibilita un consejo genético eficaz12 y, si la mutación ha sido descrita con anterioridad, permite aproximarse al posible curso de la enfermedad. A todos los portadores de la mutación de esta familia se les ha ofrecido consejo genético para planificar su descendencia. Los futuros pacientes con MCH asociada a la misma mutación en el gen MYBPC3 podrán ser informados acerca del posible curso de la enfermedad según el análisis de la familia descrita, con las limitaciones expresadas anteriormente.
No está establecido cuál debe ser el seguimiento a realizar en sujetos portadores de mutación que no han desarrollado MCH13. Un estudio reciente señala la superioridad de la resonancia magnética (RM) sobre el ecocardiograma tradicional en el diagnóstico de MCH en portadores sanos14. Los 3 portadores de la mutación sin cardiopatía de esta familia (de 19, 15 y 28 años) no presentaban signos compatibles con MCH en el estudio de RM. Para estos sujetos planificamos seguimientos con ECG, ecocardiograma y RM con periodicidad anual mientras permanezcan asintomáticos.
En resumen, este trabajo describe una mutación asociada a MCH de mal pronóstico no conocida con anterioridad, y muestra la utilidad clínica de efectuar estudio genético en pacientes con MCH.
Este trabajo ha sido financiado parcialmente gracias a una beca MAPFRE Medicina 2005.
Correspondencia: Dr. P. García-Pavía.
Servicio de Cardiología. Hospital Universitario Puerta de Hierro.
San Martín de Porres, 4. 28035 Madrid. España.
Correo electrónico: pablogpavia@yahoo.es
Recibido el 27 de marzo de 2006.
Aceptado para su publicación el 30 de agosto de 2006.