Sra. Editora:
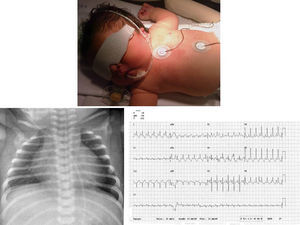
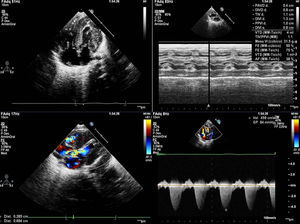
Presentamos el caso de una niña con diagnóstico intraútero en la semana 32 de gestación de miocardiopatía hipertrófica. Primer embarazo de madre añosa, feto con pliegue nucal aumentado en despistaje del primer trimestre, el cariotipo por amiocentesis fue 46 XX normal y polihidramnios. Nace a las 38 semanas de gestación sin incidencias. Se ingresa para estudio, apreciándose a la exploración física rasgos dismórficos y algo toscos, con macrocefalia y macrosomía, tórax ancho con extremidades cortas, implantación baja de las orejas, puente nasal amplio e hipertelorismo, oblicuidad ocular antimongoloide, boca en carpa con filtrum marcado y paladar ojival, implantación baja del cabello en zona posterior, y cuello corto y ancho (Figura 1) con soplo sistólico eyectivo variable desde las primeras horas de vida. El electrocardiograma (Figura 1) mostraba signos de hipertrofia biventricular con alteraciones de la repolarización. La radiografía de tórax (Figura 1) mostraba cardiomegalia moderada y el ecocardiograma (Figura 2) confirma el diagnóstico de miocardiopatía hipertrófica con severa obstrucción en tracto de salida ventricular izquierdo. Se inicia tratamiento con propranolol, permaneciendo asintomática cardiológicamente, sin arritmias documentadas. Se solicitaron estudios de despistaje de enfermedades metabólicas que fueron normales y estudio genético molecular para síndrome de Noonan que confirmó una mutación en el gen KRAS. El estudio familiar resultó negativo en los progenitores.

Figura 1. Fenotipo descrito. Cardiomegalia radiológica. Alteraciones en electrocardiograma.

Figura 2. Ecocardiografía: hipertrofia ventricular. Obstrucción severa subaórtica.
El síndrome de Noonan está asociado con gran variedad de signos y síntomas debido a la afectación multiorgánica de este. Algunos de los signos son muy característicos, como la talla baja, mientras que otros, aunque menos llamativos, pueden proporcionar la sospecha diagnóstica, como cuello corto y ancho; orejas de implantación baja y posterior, hipertelorismo con oblicuidad antimongoloide de los ojos, epicantus, ptosis, retromicrognatia, filtrum nasal ancho. También pueden presentar deformidades de tórax y espalda, hiperlaxitud articular, criptorquidia, retraso puberal, defectos de coagulación, etc. La mayoría de los pacientes tienen inteligencia normal, aunque un 15-35% presenta retraso leve y los problemas de lenguaje son comunes1.
Se han descrito mutaciones en 4 genes: PTPN11 (50%), SOS1 (20%), RAF1 (15%), KRAS (5%) y 10-15% idiopáticos2. En los casos descritos (35 en total) con la mutación KRAS el fenotipo fue más severo que el resto3.
Las anomalías cardiacas son defectos muy comunes en este síndrome y variables en cuanto a su gravedad, siendo las más frecuentes la estenosis pulmonar (60%) y la miocardiopatía hipertrófica (MCH) (20%). La MCH es una enfermedad primaria del músculo cardiaco caracterizada por hipertrofia de uno (generalmente el izquierdo) o los dos ventrículos sin una patología cardiaca o sistémica que la justifique. Aunque la mayoría de los casos son idiopáticos, en recién nacidos hay que descartar diversas patologías, siendo la más frecuente la fetopatía diabética. Puede asociarse con síndromes polimalformativos, como el de Noonan4, el de LEOPARD, el de Beckwith-Wiedemann o el de Barth, y con enfermedades metabólicas, debiendo descartar enfermedades de depósito (glucogenosis, mucopolisacaridosis, oligosacaridosis, esfingolipidosis, etc.), anomalías del metabolismo energético (déficit oxidativo de acidos grasos y déficit de carnitina), y síndromes neuromusculares (ataxia de Friedrich)5.
La edad de presentación de la MCH es muy variable y depende sobre todo de la mutación causal, aunque en muchos casos constituye parte de la evolución natural de la enfermedad, desarrollándose en edades posteriores con severidad variable6. La ecocardiografía nos da el diagnóstico definitivo, y la monitorización electrocardiográfica es necesaria para detectar precozmente la existencia de arritmias ventriculares, en algunos casos precursoras de episodios de muerte súbita de origen cardiaco (MCS), que son la principal causa de mortalidad. El riesgo de MCS es mayor a mayor severidad y a mayor precocidad de presentación, puesto que estos pacientes además presentan peores opciones terapeúticas7.
Se recomienda iniciar tratamiento farmacológico en niños asintomáticos con MCH obstructiva y gradientes significativos en el tracto de salida ventricular izquierdo y se debe realizar una adecuada estratificación del riesgo de MCS y ofrecer consejo genético a los progenitores8.
Autor para correspondencia: tonisanchan@hotmail.com