La miocardiopatía no compactada del ventrículo izquierdo (MNCVI) se caracteriza por la presencia de múltiples trabeculaciones ventriculares prominentes y recesos intertrabeculares profundos1. Se identifican antecedentes familiares en un 18-50% de los adultos y la prevalencia estimada en estudios ecocardiográficos es de un 0,014-1,300%. Su base genética es heterogénea1-3 y se han descrito solo dos mutaciones en el gen de la alfa-actina cardiaca (ACTC1)3: la ACTC1M271V y la ACTC1E101K, adicionalmente asociadas a miocardiopatía hipertrófica apical, llenado restrictivo y defectos del tabique4. Como en otros trastornos familiares hereditarios, identificar una mutación patogénica puede ser de gran utilidad para el cribado de los familiares en riesgo5.
Presentamos a una familia con MNCVI causada por la nueva mutación ACTC1I289T heterocigota. Los familiares afectados presentaron distinto curso evolutivo, diferentes manifestaciones clínicas que consistieron en MNCVI aislada, MNCVI asociada a comunicación interauricular y miocardiopatía restrictiva asociada a comunicación interauricular.
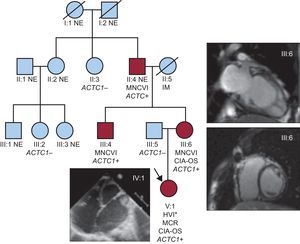
A una niña de 9 meses de edad, se le practicó un trasplante cardiaco en otro hospital debido a una miocardiopatía restrictiva con dilatación auricular, deterioro de la fracción de eyección del ventrículo izquierdo y una pequeña comunicación interauricular por ostium secundum asociada (figura, probando, IV:1). En el momento del alta se notificó la presencia de una MNCVI en la evaluación macroscópica del corazón no sospechada con anterioridad. No se realizó estudio histológico ni se conservaron muestras del corazón explantado para ulterior examen. Se ofreció a los familiares de primer grado un estudio diagnóstico detallado, aprobado por el comité local de ética de investigación, y se amplió el árbol genealógico conforme a los resultados obtenidos. Esta evaluación incluyó electrocardiograma, ecocardiografía y obtención de muestras de sangre para estudios genéticos. La realización de resonancia magnética cardiaca, prueba de esfuerzo y electrocardiograma-Holter se dejó a criterio del cardiólogo. La MNCVI se definió con los criterios de Jenni (miocardio no compactado/compactado telesistólico > 2 según la ecocardiografía) y/o de Petersen (miocardio no compactado/compactado telediastólico > 2,3 según la resonancia magnética cardiaca)1.

Árbol genealógico. ACTC1: gen de alfa-actina cardiaca; CIA-OS: comunicación interauricular de tipo ostium secundum; HVI: hipertrabeculación del ventrículo izquierdo; IM: infarto de miocardio; MCR: miocardiopatía restrictiva; MNCVI: miocardiopatía no compactada del ventrículo izquierdo; NE: no evaluado. Los círculos indican mujeres y los cuadrados, varones; los símbolos negros corresponden a individuos afectados. Esta figura se muestra a todo color solo en la versión electrónica del artículo. *Evaluación macroscópica en el momento del trasplante cardiaco.
Se llevó a cabo una secuenciación de Sanger (genes de la cadena pesada de la beta-miosina cardiaca o MYH7, proteína C de unión a la miosina C3, Nkx2.5 y ACTC1) en el ADN genómico del tío materno de la probando, puesto que inicialmente no se disponía de esta última para el estudio genético. Se identificó la mutación ACTC1I289T heterocigota y se realizó estudio genético familiar en cascada. No se identificó ninguna otra mutación en el resto de los genes analizados. En la tabla se presentan los resultados del estudio en los individuos afectados (II:4, III:4, III:6 y IV:1) y no afectados (no se pudo estudiar a ningún otro familiar).
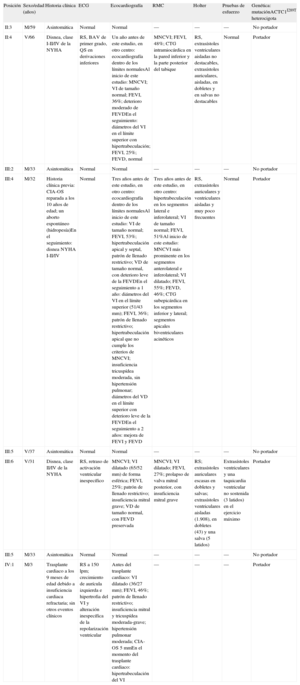
Resultados de la evaluación clínica de la familia
| Posición | Sexo/edad (años) | Historia clínica | ECG | Ecocardiografía | RMC | Holter | Pruebas de esfuerzo | Genética: mutaciónACTC1I289T heterocigota |
| II:3 | M/59 | Asintomática | Normal | Normal | — | — | — | No portador |
| II:4 | V/66 | Disnea, clase I-II/IV de la NYHA | RS, BAV de primer grado, QS en derivaciones inferiores | Un año antes de este estudio, en otro centro: ecocardiografía dentro de los límites normalesAl inicio de este estudio: MNCVI; VI de tamaño normal; FEVI, 36%; deterioro moderado de FEVDEn el seguimiento: diámetros del VI en el límite superior con hipertrabeculación; FEVI, 25%; FEVD, normal | MNCVI; FEVI, 48%; CTG intramiocárdica en la pared inferior y la parte posterior del tabique | RS, extrasístoles ventriculares aisladas no destacables, extrasístoles auriculares, aisladas, en dobletes y en salvas no destacables | Normal | Portador |
| III:2 | M/33 | Asintomática | Normal | Normal | — | — | — | No portador |
| III:4 | M/32 | Historia clínica previa: CIA-OS reparada a los 10 años de edad; un aborto espontáneo (hidropesía)En el seguimiento: disnea NYHA I-II/IV | Normal | Tres años antes de este estudio, en otro centro: ecocardiografía dentro de los límites normalesAl inicio de este estudio: VI de tamaño normal; FEVI, 53%; hipertrabeculación apical y septal, patrón de llenado restrictivo; VD de tamaño normal, con deterioro leve de la FEVDEn el seguimiento a 1 año: diámetros del VI en el límite superior (51/43 mm); FEVI, 36%; patrón de llenado restrictivo; hipertrabeculación apical que no cumple los criterios de MNCVI; insuficiencia tricuspídea moderada, sin hipertensión pulmonar; diámetros del VD en el límite superior con deterioro leve de la FEVDEn el seguimiento a 2 años: mejora de FEVI y FEVD | Tres años antes de este estudio, en otro centro: hipertrabeculación en los segmentos lateral e inferolateral; VI de tamaño normal; FEVI, 51%Al inicio de este estudio: MNCVI más prominente en los segmentos anterolateral e inferolateral; VI dilatado; FEVI, 55%; FEVD, 46%; CTG subepicárdica en los segmentos inferior y lateral; segmentos apicales biventriculares acinéticos | RS, extrasístoles auriculares y ventriculares aisladas y muy poco frecuentes | Normal | Portador |
| III:5 | V/37 | Asintomática | Normal | Normal | — | — | — | No portador |
| III:6 | V/31 | Disnea, clase II/IV de la NYHA | RS, retraso de activación ventricular inespecífico | MNCVI; VI dilatado (65/52 mm) de forma esférica; FEVI, 25%; patrón de llenado restrictivo; insuficiencia mitral grave; VD de tamaño normal, con FEVD preservada | MNCVI; VI dilatado; FEVI, 27%; prolapso de valva mitral posterior, con insuficiencia mitral grave | RS; extrasístoles auriculares escasas en dobletes y salvas; extrasístoles ventriculares aisladas (1.908), en dobletes (43) y una salva (5 latidos) | Extrasístoles ventriculares y una taquicardia ventricular no sostenida (3 latidos) en el ejercicio máximo | Portador |
| III:5 | M/33 | Asintomática | Normal | Normal | — | — | — | No portador |
| IV:1 | M/3 | Trasplante cardiaco a los 9 meses de edad debido a insuficiencia cardiaca refractaria; sin otros eventos clínicos | RS a 150 lpm; crecimiento de aurícula izquierda e hipertrofia del VI y alteración inespecífica de la repolarización ventricular | Antes del trasplante cardiaco: VI dilatado (36/27 mm); FEVI, 46%; patrón de llenado restrictivo; insuficiencia mitral y tricuspídea moderada-grave; hipertensión pulmonar moderada; CIA-OS 5mmEn el momento del trasplante cardiaco: hipertrabeculación del VI | — | — | — | Portador |
ACTC1: gen de alfa-actina cardiaca; BAV: bloqueo auriculoventricular; CIA-OS: comunicación interauricular de tipo ostium secundum; CTG: captación tardía de gadolinio; ECG: electrocardiograma; FEVD: fracción de eyección del ventrículo derecho; FEVI: fracción de eyección del ventrículo izquierdo; M: mujeres; MNCVI: miocardiopatía no compactada del ventrículo izquierdo; NYHA: New York Heart Association; RMC: resonancia magnética cardiaca; RS: ritmo sinusal; V: varones; VD: ventrículo derecho; VI: ventrículo izquierdo.
La secuenciación génica reveló la mutación ACTC1I289T heterocigota, que no constaba en la lista del National Center for Biotechnology de los polimorfismos de un solo nucleótido en el gen ACTC1. Aunque se han identificado centenares de variantes en los genes sarcoméricos y desmosómicos, se han descrito solo unos pocos polimorfismos y < 30 mutaciones en el gen ACTC1 causantes de algún tipo de miocardiopatía, lo cual indica que los cambios que se producen en este gen ACTC1 son mal tolerados. La actina es esencial para la morfología, la adhesión y la migración celular. Esta nueva variante altera un residuo aminoacídico conservado (I289) de la proteína y sustituye un aminoácido no polar (isoleucina) por otro polar y sin carga eléctrica (treonina), con lo que da lugar a modificaciones moderadas de sus propiedades fisicoquímicas relacionadas con la hidrofugacidad, la carga eléctrica, la polaridad y la masa de la proteína (distancia de Grantham, 89 [0-215]). La predicción de los análisis in silico (SIFT [Sorting Intolerant from Tolerant], Polyphen-2 y Pmut) no confirmó ni descartó su patogenicidad (resultados no concluyentes con bajo nivel de confianza). El residuo aminoacídico conservado I289 se sitúa en un subdominio 3, que es importante para la estabilidad y la polimerización de los filamentos de actina6 y está próximo al lugar de unión de la miosina, alterado posiblemente por la presencia de la mutación ACTC1I289T. Además, esta mutación ACTC1I289T mostró una cosegregación perfecta con el fenotipo de MNCVI, con una penetrancia del 100% en los individuos que se pudo estudiar.
Hay que reconocer que un estudio genético más exhaustivo podría haber incluido otros muchos genes. No obstante, se dio por terminado por lo que respecta a su relación coste-eficacia por tres razones: a) los resultados de este estudio concordaban con los de un estudio previo que relacionó la MNCVI y los defectos septales debidos a una mutación de ACTC14; b) la variante mostraba una fuerte cosegregación con el fenotipo, y c) se consideró probablemente patogénicas a las consecuencias moleculares de la variante. Otros datos funcionales obtenidos de modelos animales pueden ser útiles para confirmar el papel causal de la mutación ACTC1I289T.
En resumen, se presenta la descripción fenotípica de una familia con MNCVI causada por una nueva mutación de ACTC1I289T heterocigota con una elevada penetrancia. Es de destacar que en la literatura científica esta es la tercera mutación de la ACTC1 causante de MNCVI junto con comunicación interauricular de tipo ostium secundum en algunos de los familiares afectados.
FINANCIACIÓNEste trabajo contó con el apoyo del Instituto de Salud Carlos III (PI11/00019, CP09/00065 y RD12/0042/0029), la Generalitat Valenciana (PROMETEO 2011/027), y la Agence Nationale de la Recherche (ANR-13-BSV1-0023-03).
Damos las gracias a los pacientes que participaron en el estudio y a Biobanco La Fe por su apoyo técnico (PT13/0010/0026).