
Presentamos a 2 pacientes diagnosticados de miocardiopatías causadas por un mismo defecto genético en el ADN mitocondrial. Ambos ilustran la importancia de integrar la información clínica para poder establecer el diagnóstico y permiten discutir las características singulares del consejo reproductivo en este tipo de enfermedades genéticas.
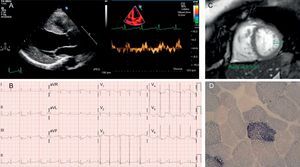
El primero es un paciente angoleño de raza negra y 47 años, trasladado a España para estudio oftalmológico. Como antecedentes destacaban diabetes mellitus de larga evolución, tuberculosis recidivante e hipoacusia bilateral neurosensorial. Ingresó por fiebre e insuficiencia respiratoria aguda. El ecocardiograma (figura) mostró un ventrículo izquierdo con hipertrofia concéntrica y disfunción sistólica grave. Se completó el estudio cardiológico con una resonancia magnética (ausencia de realce tardío) y una coronariografía (normal). Otros hallazgos fueron una llamativa caquexia, con marcha en «estepaje» e insuficiencia renal con microalbuminuria. Su madre había fallecido por problemas cardiacos, sus hijos estaban sanos y una hermana padecía una cardiopatía no especificada. Se sospechó una enfermedad mitocondrial, en concreto el síndrome MIDD (sordera y diabetes mellitus de herencia materna) (tabla). La secuenciación del ADN mitocondrial a partir de muestra sanguínea documentó la mutación m.3243A>G en el gen MT-TL1 que codifica para tRNALeu mitocondrial con un grado de heteroplasmia del 50%. El paciente fue dado de alta con tratamiento para insuficiencia cardiaca y regresó a su país.

A: ecocardiograma del caso 1 que muestra un ventrículo izquierdo hipertrófico y dilatado, con disminución de las velocidades del Doppler tisular. B: electrocardiograma del caso 1 en ritmo sinusal, con signos de crecimiento ventricular izquierdo. C: resonancia magnética cardiaca del caso 2, con aumento de la trabeculación y relación no compactado/compactado > 2,3. D: fibras rojas rasgadas con hipertinción con succinato deshidrogenasa en el caso 2.
Principales síndromes mitocondriales asociados a la mutación m.3243A>G en MT-TL1 que cursan con afección cardiaca
| Síndrome | Clínica | Afección cardiaca | Genética |
|---|---|---|---|
| MELAS | Encefalomiopatía, acidosis láctica y episodios tipo ictus; epilepsia; vómitos; cefaleas; talla baja; hipoacusia | Miocardiopatía (MCH, MCD, MCNC, MCR) y arritmias (WPW y BAV) | Mitocondrial (el 80% m.3243A>G y el 10% m.3271T>C en tRNALeu) |
| MIDD | Sordera y DM, retinopatía, miopatía, miocardiopatía, talla baja, insuficiencia renal | Miocardiopatía (MCH) y arritmias | Mitocondrial (> 85% m.3243A>G en tRNALeu) y deleciones múltiples |
| MERRF | Epilepsia mioclónica y fibras rojas rasgadas; demencia, atrofia óptica, hipoacusia, neuropatía periférica | Miocardiopatía (MCH) y arritmias (WPW y trastornos de la conducción) | Mitocondrial (el 80% m.8344A>G en tRNALys; m.3243A>G en tRNALeu en casos aislados) |
| Síndrome de Kearns-Sayre | Oftalmoplejía externa crónica progresiva, retinosis pigmentaria con/sin miopatía | Trastornos de la conducción (BAV) y miocardiopatías (MCD, MCR) | Mitocondrial (deleción: lo más común; m.3243A>G en tRNALeu en casos aislados) |
| Síndrome de Leigh | Encefalomielopatía necrosante subaguda; retraso psicomotor, ataxia, crisis convulsivas, oftalmoplejía y acidosis láctica | Miocardiopatía (MCH) y arritmias | Nuclear o mitocondrial (mutaciones en tARNVal; tRNALeu en algunos casos) |
BAV: bloqueo auriculoventricular; CPEO: oftalmoplejía externa crónica progresiva; DM: diabetes mellitus; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MCNC: miocardiopatía no compactada: MCR: miocardiopatía restrictiva; MELAS: encefalopatía mitocondrial, acidosis láctica y episodios similares a ictus; MERRF: epilepsia mioclónica con fibras rojas rasgadas; MIDD: DM y sordera de herencia materna; WPW: síndrome de Wolff-Parkinson-White.
El segundo caso es una mujer de 36 años con múltiples microinfartos cerebrales identificados en un estudio por hipoacusia a la que, tras valoración cardiológica, se le diagnosticó una posible miocardiopatía no compactada (figura). La paciente era diabética insulinodependiente desde los 24 años y presentaba talla e índice de masa corporal bajos. No tenía antecedentes familiares de cardiopatía, miopatía o problemas neurosensoriales. Refería migrañas frecuentes. Estaba en clase funcional de la New York Heart AssociationII-III por intolerancia al ejercicio, con creatincinasa y fracción aminoterminal del propéptido natriurético cerebral normales y cifras oscilantes de lactato (> 2,5 mmol/l en varias determinaciones). No se pautó ningún tratamiento adicional, ya que la función ventricular era normal. En este caso, el fenotipo clínico sospechado fue el síndrome MELAS (encefalomiopatía, acidosis láctica y episodios parecidos a ictus) (tabla). La biopsia muscular mostró un 8% de fibras rojas rasgadas, la mayoría positivas a ciclooxigenasa (figura), y el estudio genético reveló nuevamente la mutación m.3243A>G en MT-TL1, con heteroplasmia del 87-91% en la biopsia muscular.
Las enfermedades mitocondriales se caracterizan por una disfunción de la cadena respiratoria que lleva a un déficit energético celular. Los órganos más afectados son aquellos con mayor demanda metabólica, como sistema nervioso o músculo. Son manifestaciones comunes encefalopatía, miopatía, diabetes mellitus e hipoacusia. La afección simultánea de varios de estos órganos sin una aparente causa común y manifestaciones a edades tempranas, como la diabetes mellitus o el ictus antes de los 40 años, deben orientar la sospecha diagnóstica. Estos síndromes pueden cursar con alteraciones cardiacas en porcentajes muy variables (del 3 al 81% según las series) y en forma tanto de miocardiopatías (hipertrófica o dilatada habitualmente) como de trastornos de conducción (tabla). La mutación m.3243A>G en el tRNALeu mitocondrial es una de las más frecuentes y puede dar lugar a síndromes diferentes como MIDD (caso 1) y MELAS (caso 2) con gran variabilidad en las manifestaciones cardiacas1. Es típico el inicio o empeoramiento de los síntomas tras situaciones de estrés (en el primer caso, la hipoacusia atribuida a tuberculostáticos realmente se había iniciado años antes, tras un episodio de paludismo). En presencia de un cuadro sindrómico clásico, el diagnóstico se confirma con el estudio genético. El tratamiento es sintomático, y se debe evitar fármacos como la metformina (riesgo de acidosis láctica) o las estatinas (empeoramiento de la miopatía). Los antioxidantes o terapias sustitutivas con coenzima Q10 o L-carnitina se emplean, aunque hay controversia en cuanto a su efecto beneficioso. Se debe tener especial precaución con la anestesia por el riesgo de fallo respiratorio y evitar los relajantes musculares no despolarizantes y los barbitúricos.
Tras el diagnóstico, la paciente del caso 2 expresó su deseo de tener hijos que no padecieran la enfermedad. Prestar consejo genético es una de las tareas a las que se enfrentan los cardiólogos que atienden a pacientes con cardiopatías familiares2.
La genética mitocondrial tiene una serie de peculiaridades que se han de tener en cuenta a la hora del consejo reproductivo. La herencia es matrilineal, la trasmiten las mujeres a toda su descendencia, ya que las mitocondrias del cigoto proceden del ovocito. La coexistencia de moléculas de ADN mitocondrial distintas se denomina heteroplasmia. Es necesario un porcentaje mínimo de ADN mutado para que aparezcan síntomas (efecto umbral). En la división celular, el reparto de mitocondrias es aleatorio y las células hijas no necesariamente reciben la misma carga de ADN mutado. El ADN mitocondrial se sigue replicando independientemente de la división celular, por lo que tejidos inicialmente sanos podrían desarrollar signos de la enfermedad con el tiempo. Todos estos aspectos explican la variabilidad fenotípica y de la expresividad clínica de estos trastornos, así como las dificultades para prevenirlos mediante técnicas de reproducción asistida. Las opciones reproductivas que garantizan la ausencia de trasmisión de la enfermedad a la descendencia son la adopción, la gestación de un embrión de otra pareja o la fecundación con un ovocito de donante. Todas ellas tienen el inconveniente de que se pierde el vínculo genético con la madre3.
El diagnóstico preimplantacional en los trastornos genéticos mitocondriales solo disminuye las posibilidades de que se produzca la enfermedad trasfiriendo embriones con bajo grado de heteroplasmia. Con la legislación española vigente, es improbable que se hubiera autorizado este tratamiento, ya que no se asegura el empleo de embriones sin defecto genético4.
Aunque todavía están en fase de experimentación, las técnicas de reemplazo mitocondrial son el futuro en la prevención de estas enfermedades. Consisten en crear embriones con ADN nuclear de los progenitores y ADN mitocondrial de una donante («bebés de 3 padres»). Un reciente documento científico británico avala la eficacia y la seguridad de estas técnicas y aboga por su traslación a humanos, pues actualmente no es posible el uso de embriones cuyo material genético haya sido modificado. Tras la publicación de este documento, y tras una consulta popular en la que la opinión pública se mostró favorable a estas técnicas, el gobierno de Reino Unido se ha planteado modificar su legislación5.
La paciente descrita finalmente decidió someterse a una fecundación in vitro con ovocito donado.
FINANCIACIÓNEste trabajo se ha realizado parcialmente gracias a financiación del ISCIII (PI11/0699 y RD12/0042/0066 a PGP y LAP; PI12/01795 a BB).