Palabras clave
INTRODUCCIÓN
El mixoma es eltumor cardiaco primario más frecuente y ha sidodiagnosticado desde recién nacidos hasta los 95 añosde edad. Sus manifestaciones clínicas pueden serembólicas, de obstrucción al vaciamiento auricularizquierdo (disnea, ortopnea) y síntomas generales comofatiga, mialgias, fiebre o pérdida de peso1,2. Seha identificado a pacientes con mixomas recurrentes que puedenafectar a varios miembros de la familia y se acompañan dealteraciones en la pigmentación de la piel e hiperactividadendocrina; estas alteraciones se conocen como complejo deCarney3.
De enero de 1986 anoviembre de 2005 estudiamos a 63 pacientes con diagnósticoecocardiográfico, quirúrgico e histopatológicode mixoma cardiaco. En 5 (7,9%) de ellos hubo recidiva del tumor.En 3 de estos 5 pacientes y en 11 de los 58 restantes seefectuó estudio genético. El propósito de estetrabajo es describir las características clínicas yecocardiográficas de los pacientes con mixomas recurrentes ylas alteraciones genéticas detectadas y compararlas con losdatos clínicos, ecocardiográficos y genéticosde los 11 pacientes con mixoma único (tabla 1).

MÉTODOS
El ADNgenómico fue extraído a partir de 10 ml de sangreperiférica utilizando técnicasconvencionales4.
Los 12 exones delgen PRKAR1A del cromosoma 17 fueron analizados por reacción en cadena dela polimerasa-polimorfismo conformacional de una sola cadena(PCR-SSCP) con el fin de detectar las regiones con posiblesmutaciones. Posteriormente estas regiones fueron secuenciadasutilizando un secuenciador automático de capilar. Lassecuencias fueron comparadas con la secuencia consenso y conaquéllas que contienen mutaciones ya comunicadas en laliteratura. Cuando apareció una mutación, serealizó un análisis codón por codónpara establecer el tipo de mutación, así como suposición en el gen5,6.
Los criterios paraestablecer el diagnóstico de complejo de Carney fueron lospropuestos por Stratakis7.
El primeroconsiste en identificar dos de las siguientes alteraciones: manchaspigmentarias de la piel, tumores de localización diversa yenfermedad adrenocortical nodular pigmentaria. El segundo criterioes tener una de las alteraciones mencionadas más laafección de un familiar de primer grado o tener unamutación en el gen PRKAR1A.
Caso 1
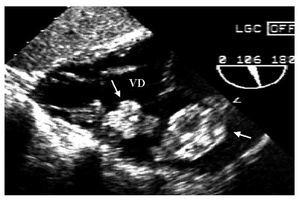
Mujer de 29años con palpitaciones y disnea. El ecocardiogramamostró dos tumoraciones en el ventrículo derecho(fig. 1), que fueron resecadas quirúrgicamente. Su estudiohistopatológico confirmó el diagnóstico demixomas. Nueve meses después, un ecocardiograma de controlmostró una masa tumoral en la misma localización. Suestudio genético fue normal.

Fig.1. Ecocardiograma transgástrico. Se observan dosmixomas (flechas) en el ventrículo derecho (VD).
Caso 2
Mujer de 18años con antecedente de amaurosis izquierda yhemiplejía derecha 3 años antes. En laexploración física se encontró un soplocardiaco y una tomografía computarizada evidencióinfartos cerebrales múltiples. El ecocardiogramatranstorácico mostró un mixoma en la porciónsuperior de la aurícula derecha y otro en la aurículaizquierda adosado al tabique interauricular. Tres añosdespués se detectaron 3 mixomas, uno pediculado y cercano ala orejuela izquierda, otro en el tracto de salida delventrículo izquierdo y un tercero en el ventrículoderecho a nivel del septo trabecular. Después de suextirpación, la paciente murió en el postoperatoriopor insuficiencia de múltiples órganos. El estudiohistopatológico de los tumores confirmó eldiagnóstico de mixomas. No fue posible realizar el estudiogenético de la paciente y sus familiares, por lo que eldiagnóstico de Carney quedó como posible.
Caso 3
Varón de 42años con cuatro hermanos, dos de ellos con lentiginosis enlos labios. A los 28 años se le diagnosticósíndrome de Cushing y léntigo en la cara y las manos.Unos meses después, se efectuó adrenalectomíabilateral con el diagnóstico de hiperplasia nodularadrenocortical pigmentaria. Al año siguiente, unecocardiograma mostró un tumor de 3,5 ´ 2,5 cm en la regiónposterobasal del ventrículo izquierdo. Se realizóresección quirúrgica y el estudiohistopatológico estableció el diagnóstico demixoma cardiaco. A los 3 años un ecocardiograma de controlmostró un nuevo mixoma ventricular izquierdo en unalocalización diferente de la previa, y el tumor fueextirpado. No fue posible efectuar estudio genético delpaciente y sus familiares; sin embargo, se cumplieron los criteriospara establecer el diagnóstico de complejo deCarney.
Caso 4
Varón de 39años con antecedentes de hemiparesia fasciocorporal derechaa los 12 y los 18 años, embolia en la pierna derecha einfarto del miocardio en la pared inferior. El ecocardiogramamostró un mixoma en la aurícula izquierda de 5´ 3 cm con tres sitios deimplantación que incluían el tabique interauricular.A los 14 años de la resección tumoral, se observanvarias masas tumorales en la aurícula izquierda, unasadheridas al tabique interauricular y otras con localizacióndiferente. Los mixomas fueron resecados y 7 añosdespués no hay evidencia de recidivas. El estudiogenético fue normal en tres hermanos, pero en el paciente yen su madre apareció una mutación que corresponde auna deleción de una base en el intrón 2 delgen PRKAR1A. Actualmente se están realizando los estudios para establecerel papel funcional de dicha mutación. Según loscriterios mencionados, se diagnosticó complejo deCarney.
Caso 5
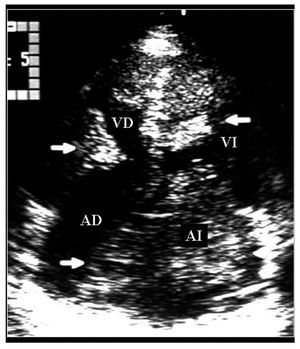
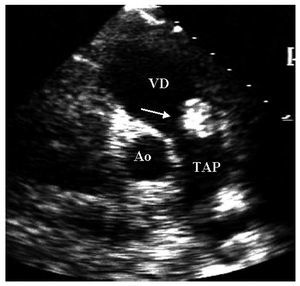
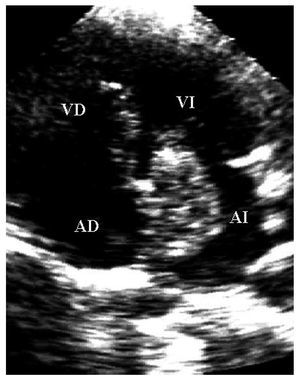
Mujer de 36años con antecedente de soplo cardiaco desde los 23años y lesiones cutáneas pigmentadas. Elecocardiograma transtorácico mostró tumores deaspecto mixoide en las 4 cavidades cardiacas8 (fig. 2).La resección completa de los mixomas fue corroborada con unecocardiograma transesofágico. Los ecocardiogramasefectuados a sus padres y cinco hermanos no mostraron masascardiacas. Dos años después requiere cirugíapor la aparición de tres nuevos mixomas localizados en ambasaurículas y en el ventrículo derecho. Ochoaños después de la segunda cirugía, seefectuó la resección quirúrgica de nuevasrecidivas tumorales localizadas en la aurícula izquierda, suorejuela y ambos ventrículos. A los 7 años de laúltima cirugía, se ha demostrado una pequeñatumoración en la vía de salida del ventrículoderecho (fig. 3). La exploración física y losecocardiogramas de sus padres y de tres hermanos fueron normales.También se estudio a sus tres hijos, y en el menor de 6años no se detectaron anormalidades relacionados con elsíndrome de Carney. En el segundo, de 8 años, elecocardiograma fue normal. Sin embargo, se detectó unapequeña tumoración en el escroto, quecorrespondió a un angiofibroma mixoide. En el tercer hijo,de 12 años, el ecocardiograma mostró un mixoma en laaurícula izquierda de 39 ´ 30 mm, adherido al tabiqueinterauricular, que se resecó (fig. 4). El estudiogenético no mostró alteraciones en los padres, treshermanos y un hijo. Tanto la paciente como dos de sus hijosmostraron una alteración en el gen PRKAR1A, consistente endeleción de una base en la posición 552 (552del g),que corresponde al codón 155 en el exón 4B. Ladeleción modifica el marco de lectura generando uncodón de paro y, por lo tanto, una proteínaincompleta y no funcional. Esto confirmó eldiagnóstico de complejo de Carney.

Fig.2. Ecocardiograma transtorácico apical que muestravarios mixomas en las cuatro cavidades cardiacas (fechas). AD:aurícula derecha; Al: aurícula izquierda; VD:ventrículo derecho; VI: ventrículoizquierdo.

Fig.3. Estudio transtorácico en el plano transversal. Laflecha muestra un mixoma en el tracto de salida delventrículo derecho.

Fig.4. Estudio apical de las cuatro cámaras. Se observa unmixoma en la aurícula izquierda con inserción en eltabique interauricular.
RESULTADOS
El grupo conmixoma cardiaco único se formó con 9 mujeres y 2varones entre los 25 y los 56 (media, 48) años de edad. Lalocalización del mixoma fue la aurícula izquierda en9 pacientes y la aurícula derecha en 2. Dos de los 11pacientes tenían el antecedente de un cuadro embólicocerebral. El seguimiento tras la resección quirúrgicadel mixoma varió de 1 a 17 (media, 6,3) años, y losregistros ecocardiográficos no mostraron recidivastumorales. El estudio genético realizado en los 11 pacientesno mostró alteraciones (tabla 1).
DISCUSIÓN
En 58 de los 63pacientes estudiados, el tumor fue único conlocalización en la aurícula izquierda (50 casos), laaurícula derecha (5) o los ventrículos (3). En 11 fueposible efectuar estudio genético, que no detectóanormalidades. No hubo signos relacionados con el complejo deCarney y su comportamiento clínico fue el habitual en elmixoma único.
El análisisde los 5 pacientes con mixomas recurrentes nos permite lossiguientes comentarios. En el caso 1, a pesar de la rareza de lalocalización tumoral, el estudio histopatológicoestableció el diagnóstico de mixomas. Su recurrenciacon la misma localización indica que su extirpaciónquirúrgica fue incompleta. En el paciente 2, eldiagnóstico de complejo de Carney no se puede descartardebido a los mixomas recurrentes en diferentes localizaciones. Enel caso 3, el diagnóstico de complejo de Carney se establecepor la presencia de léntigo, hiperplasia nodular corticalpigmentaria y un mixoma recidivante en el ventrículoizquierdo. Dos de sus hermanos también teníanalteración en la pigmentación cutánea. En elpaciente 4, el diagnóstico de complejo de Carney seefectuó porque concurrían mixomas recurrentes dediferente localización y alteraciones en el gen PRKAR1Aen el paciente y sumadre. En la paciente 5, el complejo de Carney se confirmópor la lentiginosis, los mixomas recurrentes y las alteracionesgenéticas en ella y dos de sus hijos; además, en unode ellos se resecó un mixoma auricular izquierdo.
En nuestra serie,el complejo de Carney representa un 4,7-6,3% de los mixomascardiacos. Además de la recurrencia observada, los mixomassuelen ser múltiples y localizados en cualquiera de lascuatro cavidades. Sin embargo, como observamos en uno de los hijosde la paciente 5, la clásica localización de un tumorúnico en la aurícula izquierda no excluye eldiagnóstico. El desarrollo de mixomas cardiacos en estesíndrome es más temprano, y en el hijo de la paciente5, fue a los 12 años. En el grupo con mixoma recurrente, elpromedio de edad fue 32 años y en el grupo con mixomaaislado, 48 años.
En cuanto aembolias arteriales, en el grupo con mixoma auricular únicola incidencia fue del 18%, cercana a la comunicadapreviamente9. En el grupo con mixomas recurrentes fuedel 40%, y es probable que en el paciente 4 el infarto delmiocardio fuera secundario a embolia coronaria.
El complejo deCarney se hereda con un patrón autosómico dominante.El análisis de ligamiento en familias con estaanomalía ha mostrado heterogeneidad genética con almenos dos loci principales como genes candidatos. Inicialmente seidentificó un locus en el cromosoma 2(2p15-16)10; sin embargo, no se ha identificado en esaregión el gen que causa el complejo de Carney. En el segundolocus, que se encuentra en el cromosoma 17(17q22-24)11,12, se identificó recientemente elgen PRKAR1A13. Este gen codifica la subunidad reguladora 1-alfade la proteincinasa A (PKA), el principal mediador de laseñalización de adenosinmonofosfato cíclico enlos mamíferos14. Se ha detectado expresiónalterada de este gen en muchos tumores de apariciónesporádica y en líneas celulares derivadas de tumoren pacientes sin complejo de Carney, y se piensa que es un gencandidato para la tumorogénesis endocrina y no endocrina enel complejo de Carney15,16.
En 2 de los 5pacientes con mixomas recidivantes, encontramos alteraciones en elgen PRKAR1A. El hecho de que no se detectaran mutaciones en el gen PRKAR1Aen individuos conmixomas únicos indica que las alteraciones de este gen serelacionan directamente con el complejo de Carney y no con laaparición de mixomas. Esto es de utilidad clínica, yaque estas alteraciones genéticas en pacientes con mixomasúnicos con localizaciones atípicas, mixomasmúltiples o con múltiples localizaciones puede ayudara establecer de forma temprana el diagnóstico de complejo deCarney. Una vez establecido el diagnóstico, se puede ampliarel estudio genético de los familiares para despuésrealizar estudios de imagen en los que tengan la mutación.Esto a su vez permitiría llevar a cabo un programa deseguimiento clínico de los pacientes y los familiares quepresenten las mutaciones, dado el riesgo de recurrencias quetienen. Sin embargo, es importante señalar que ladetección de mutaciones no ayuda en el caso de recidivas porresección incompleta del mixoma, que se tendrá quedetectar por los métodos convencionales. También esimportante considerar que el complejo de Carney es unaalteración con heterogeneidad genética, por lo que nodetectar mutaciones en el gen PRKAR1A en los pacientes nodescarta que otro gen esté participando en su desarrollo;como ya se ha mencionado, otro gen causal podría estarubicado en el cromosoma 2.
Entre laslimitaciones de este estudio, debemos mencionar que sudiseño fue retrospectivo y observacional; por lo pocofrecuente de la afección estudiada, la evaluación delos pacientes sólo fue descriptiva y analítica. Encuanto a la alteración genética detectada en el caso4, está pendiente el resultado del estudio orientado paraestablecer el papel funcional de la mutación encontrada. Eneste sentido, el estudio genético se extenderá alresto de los 63 pacientes que no se incluyeron en este estudio yasisten a la consulta externa del hospital.
Por los resultadosobtenidos, podemos señalar las siguientesconclusiones: a) se debe investigar un posible complejo de Carney en todos lospacientes con mixomas intracardiacos; b) el complejo de Carney esmás frecuente en pacientes con mixomas recurrentes orecidivantes, y además es común que los tumoresocupen dos o más cavidades cardiacas, y c) la detección demutaciones en pacientes con mixomas únicos delocalización atípica o mixomas múltiples,así como en sus familiares, podría ser de utilidadpara establecer un diagnóstico temprano de la enfermedad yasí llevar un seguimiento clínico que permitadetectar recurrencias en estos individuos.
Full English text available from: www.revespcardiol.org
Correspondencia: Dr. J. Vargas-Barrón.
Departamento de Ecocardiografía. Instituto Nacional deCardiología Ignacio Chávez.
Juan Badiano, 1. Col. Sección XVI. 14080 Tlalpan.México DF. México. Correo electrónico: eco_vargas@terra.com.mx
Recibido el7 de septiembre de 2007.
Aceptado para su publicación el 22 de enero de2008.