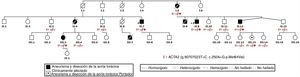

Los aneurismas de aorta torácica no sindrómicos (AATNS)1 se caracterizan por la formación silente de aneurismas y disecciones de aorta sin otras manifestaciones externas que ayuden a su diagnóstico. En los de origen familiar, se ha descrito una herencia autosómica dominante, con penetrancia incompleta y expresión variable2, identificándose mutaciones en diversos genes, las más frecuentes localizadas en el gen ACTA23–5. Se presenta el caso de una familia en la que se encontró una nueva mutación en ACTA2, no descrita previamente, causante de AATNS.
El caso índice fue un varón (II.9), intervenido a los 50 años de disección de aorta (DA) tipo A, con varios familiares fallecidos de muerte súbita (figura 1, tabla 1). Su madre (I.5) presentaba dilatación de raíz aórtica (RA) de 38 mm, y su hermano (II.8) falleció por DA abdominal. Los otros 4 hermanos estaban aparentemente sanos. Se completó el pedigrí con 2 primas hermanas, fallecidas de muerte súbita, una (II.6) a los 45 años sin autopsia (un hijo de esta falleció a los 17 años por DA confirmada por autopsia), y otra (II.4) por DA ascendente a los 47.

Descripción de los familiares estudiados
| Paciente | I.5 | II.3 | II.4 | II.5 | II.7 | II.10 | II.11 | II.12 | II.13 | III.3 | III.4 | III.6 | III.10 | III.11 | III.12 | III.13 | III.14 | III.15 | III.16 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Edad 1.a evaluación | 83 | 47 | 43 | 41 | 36 | 45 | 42 | 39 | 39 | 27 | 29 | 20 | 16 | 11 | 21 | 15 | 12 | 20 | 14 |
| Diámetro 1.a evaluación (mm) | 38 | 36 | 38 | 39 | 40 | 29 | 31 | 37 | 38 | 39 | 30 | 23 | 31 | Normal | 24 | 27 | Normal | 32 | 23 |
| Z-core, última revisión | 2,31 | 1,55 | 3,67 | 1,24 | 4,42 | –0,93 | 0,61 | 2,64 | 1,22 | 2,43 | 0,64 | –1,04 | 1,44 | Normal | –0,93 | –0,23 | Normal | 0,33 | 0,45 |
| Ritmo de crecimiento (mm/año) | 1 | 0,6 | 1,5 | 0,2 | 0,8 | 0,2 | 1 | 0,6 | 0 | 0,3 | 0 | 0,8 | – | – | – | – | – | – | – |
| Técnica de imagen | EcoAngio-TC | EcoAngio-RM | EcoAngio-TC | EcoAngio-TC | EcoAngio-TC | Eco | Eco | EcoAngio-TC | Eco | EcoAngio-RM | Eco | Eco | Eco | Eco | Eco | Eco | Eco | Eco | Eco |
| Evento/cirugía preventiva | No | No | Sí | No | Sí | No | No | No | No | No | No | No | No | No | No | No | No | No | No |
| ACTA2 | + | – | NR | – | + | – | – | + | – | + | – | NP | NP | NP | – | + | – | NP | NP |
Angio-RC: angiografía por resonancia magnética; Angio-TC: angiografía por tomografía computarizada; NP: no procede; NR: no realizado.
Se realizó estudio genético mediante secuenciación de nueva generación de un panel que incluyó 41 genes relacionados con enfermedad aórtica (ACTA2, ADAMTSL4, B3GAT3, CBS, COL1A1-2, COL3A1, COL5A1-2, EFEMP2, ELN, FBN1-2, FLNA, GAA, GATA5, HRAS, KCNJ8, MED12, MYH11, MYLK, NKX2-5, NOTCH1, PLOD1, PRKG1, PTPN11, SKI, SLC2A10, SMAD3-4, TGFB2-3, TGFBR12, ZDHHC9, ATP7A, CHST14, ADAMTS2*, B4GALT7*, FKBP14*, SLC39A13*), y se identificó una mutación en heterocigosis en el exón 3 del gen ACTA2, que codifica la proteína alfa actina de músculo liso, no descrita previamente: Met84Val (NC_000010.10:g.90707023T>C). Posteriormente se examinó a un total de 19 familiares; a 13 se les realizó estudio genético. En 4 de ellos se evidenció dilatación de RA, todos con estudio genético positivo. Se estudió a todos los familiares mediante ecocardiografía y se completó el estudio de los portadores afectados con angiografía por tomografía computarizada o angiografía por resonancia magnética. La mediana de edad de presentación fue 47 años, y solo 1 miembro de la familia tuvo un comienzo precoz (17 años). En nuestra serie, la localización predominante de dilatación fue la RA, y apareció 1 solo caso de aneurisma abdominal.
Se recomendó a los pacientes control estricto de factores de riesgo cardiovascular y evitar ejercicios isométricos y de competición. Todos los casos fueron tratados con antagonistas del receptor de la angiotensina II, y 1 de ellos se derivó a cirugía profiláctica.
La DA en pacientes jóvenes es a menudo el resultado de una aortopatía genética, y el síndrome de Marfan es su paradigma. Existen otras aortopatías genéticas carentes de signos externos, caracterizadas por la formación silente de aneurismas de aorta torácica y disecciones, conocidos como AATNS1. Las mutaciones identificadas en el gen ACTA2 explicarían el 14% de estos casos3–5. El gen ACTA2, localizado en el cromosoma 10 (10q23.31), codifica la alfa actina, la proteína más abundante del músculo liso de las células vasculares. Su déficit origina disminución de la contractilidad de estas células, y puede provocar DA tipo A y B4,5. La variante identificada en esta familia no ha sido descrita previamente en la literatura, ni en bases de datos públicas de genotipado en población general, además afecta a un residuo altamente conservado. Se han identificado variantes en aminoácidos cercanos, como p.Asp82Glu y p.Glu85Lys, asociadas con el desarrollo de enfermedad de la aorta torácica. Los AATNS familiares presentan una expresión variable2. En esta familia la mediana de edad de presentación fue 47 años, similar a la descrita. Otra característica de la variabilidad fenotípica del AATNS es el nivel de la afección aórtica; en nuestro caso predomina a nivel de RA. Está descrito que la penetrancia es incompleta; en nuestro caso, de los 6 pacientes con estudio genético positivo, 5 presentaron dilatación de RA, lo que indica que la variante identificada probablemente tenga elevada penetrancia. Aunque se ha descrito que la penetrancia es menor en mujeres, en esta familia se describieron casos en ambos sexos. Otras mutaciones descritas en ACTA2 se asociaron con livedo reticularis, iris flocculi y válvula aórtica bicúspide4; sin embargo, estas entidades no estaban presentes en ninguno de nuestros pacientes, lo que puede indicar baja frecuencia de asociación, como ya se había señalado en estudios previos4.
El tratamiento consiste en abordar el estrés hemodinámico mediante fármacos para prevenir complicaciones o reparación quirúrgica. Las indicaciones de cirugía profiláctica no están claramente establecidas6, aunque parece razonable indicarla ante diámetros aórticos> 5cm o incluso menores (> 4,5cm), o con tasas de crecimiento superiores a 0,5cm/año. En nuestra familia solo se ha descrito 1 caso de disección con RA de 42 mm (ecocardiograma realizado 9 meses antes). Medidas preventivas, como controlar la hipertensión y evitar el tabaco, el ejercicio isométrico y los deportes competitivos, podrían tener un impacto en la evolución de la enfermedad6.
Las mutaciones en ACTA2 son la causa más frecuente de AATNS familiares. Se describe una nueva mutación en el gen ACTA2 que cosegrega en una gran familia. En esta mutación, la dilatación aparece fundamentalmente a nivel de la RA, y pueden producirse eventos con diámetros ligeramente aumentados. Son importantes el diagnóstico precoz de familiares (clínico-genético-imagen), su seguimiento (clínico-imagen) y la prevención de complicaciones (control de factores de riesgo cardiovascular).
CONFLICTO DE INTERESESL. Montserrat es accionista de Health in Code S.L.