
La miocardiopatía hipertrófica (MCH) es un trastorno autosómico dominante y es la cardiopatía hereditaria más frecuente1. Aunque son varias las mutaciones relacionadas con la MCH de mal pronóstico, el valor pronóstico de mutaciones concretas en la MCH sigue siendo controvertido. Las variantes genéticas de la cadena pesada de la β-miosina codificadas por MYH7 que involucran la llamada «región conversora» se han asociado con desarrollo temprano de la enfermedad, con mayor incidencia de arritmias ventriculares malignas y aumento de la necesidad de trasplante cardiaco2. La región conversora se extiende de Phe-709 a Arg-777 y se cree que es un elemento clave del complejo actina-miosina donde tiene lugar la deformación elástica.
A continuación se describe el caso de una familia con una variante genética no descrita previamente en la región conversora del gen MYH7 (c.2212A > C; p.Ser738Arg). El caso índice es una mujer de 42 años a la que se diagnosticó MCH cuando tenía 24 años y que ingresó en planta de cardiología a causa de un episodio de insuficiencia cardiaca. El ecocardiograma mostró disfunción sistólica grave de ambos ventrículos. Aunque la paciente recibió un tratamiento específico para la insuficiencia cardiaca (tabla 1), a los 45 años requirió un trasplante cardiaco.
Características clínicas de los pacientes
| Paciente | Año de nacimiento | Edad en el diagnóstico de MCH, años | Grosor miocárdico máximo | Gradiente del TSVI | Insuficiencia mitral | FEVI | RTG | Síntomas | Arritmia | Tratamiento | Dispositivos implantables | Resultado final |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(caso índice) | 1960 | 24 años(1984) | > 18 mm | ND | Moderada (funcional) | < 30% | ND | Síncope(29 años)Insuficiencia cardiaca(42 años) | Fibrilación auricular | IECASotalolDigitalisWarfarinaDiuréticos del asa | DAI-TRC(2002) | Trasplante(45 años) |
| 2 | 1990 | 22 años(2012) | 18 mm | 48 mmHg | Moderada(SAM) | 61% | Sí | Ninguno | TVNS | Atenolol | DAI(2015) | Seguimiento |
DAI: desfibrilador automático implantable; FEVI: fracción de eyección del ventrículo izquierdo; IECA: inhibidor de la enzima de conversión de la angiotensina; MAS: movimiento anterior sistólico; MCH: miocardiopatía hipertrófica; ND: no disponible; RTG: realce tardío de gadolinio; TRC: terapia de resincronización cardiaca; TSVI: tracto de salida del ventrículo izquierdo; TVNS: taquicardia ventricular no sostenida.
Se realizó un test genético con secuenciación de nueva generación con un panel general de miocardiopatías en el que se incluían 17 genes (ACTC, DES, FLNC, GLA, LAMP2, MYBPC3, MYH7, MYL2, MYL3, PLN, PRKAG2, PTPN11, TNNC1, TNNI3, TNNT2, TPM1 y TTR) con un secuenciador Illumina Hiseq. La alineación y el filtrado de las variantes se realizó en un pipeline propio. La patogenicidad de las variantes se clasificó según las recomendaciones del American College of Medical Genetics and Genomics.
Se identificó una variante genética tipo cambio de sentido en el gen MYH7 (c.2212A > C; p.Ser738Arg). No se encontró descripción o información alguna de esta variante en las bases de datos de la población general. La variante afecta a un aminoácido conservado de la región conversora de la proteína. Se han descrito otras 3 variantes en el mismo residuo asociadas con MCH (p.Ser738Asn, p.Ser738Arg y p.Ser738Thr). Los predictores in silico (SIFT, Polyphen-2) la consideran probablemente deletérea. La variante se consideró probablemente patogénica.
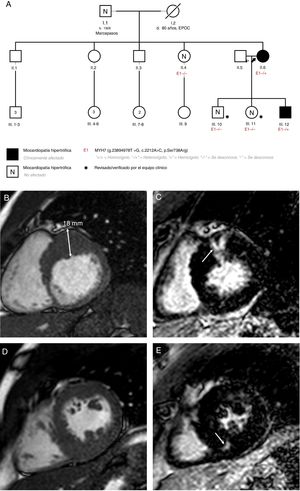
El estudio familiar mostró que la paciente tenía 2 hijos sanos (un varón y una mujer) que no son portadores de la variante. El tercer hijo, un varón al que se diagnosticó MCH cuando tenía 22 años, era portador. La madre de la paciente ya había fallecido cuando a esta se le diagnóstico la MCH. Su padre está vivo y se ha descartado la existencia de MCH con ecocardiograma, aunque rehusó dar el consentimiento para el estudio genético. En una de las hermanas de la paciente, la MCH se descartó tras una evaluación completa y pruebas genéticas. Los otros 3 hermanos se negaron a someterse a estudio. El estudio genealógico (pedigree) se muestra en la figura 1A.
. D: caso 2, resonancia magnética cardiaca; músculos papilares en eje corto. E: caso 2, resonancia magnética cardiaca; realce tardío de gadolinio en el segmento medio inferoseptal (flecha). EPOC: enfermedad pulmonar obstructiva crónica; TxC: trasplante cardiaco.")
A: estudio genealógico de los pacientes; la flecha negra apunta al caso índice. B: caso 2, resonancia magnética cardiaca, eje corto basal. C: caso 2, resonancia magnética cardiaca; realce tardío de gadolinio en el segmento basal anterior (flecha). D: caso 2, resonancia magnética cardiaca; músculos papilares en eje corto. E: caso 2, resonancia magnética cardiaca; realce tardío de gadolinio en el segmento medio inferoseptal (flecha). EPOC: enfermedad pulmonar obstructiva crónica; TxC: trasplante cardiaco.
El hijo afectado actualmente tiene 28 años. Se ha documentado hipertrofia grave (grosor máximo de la pared de 18 mm) con función ventricular normal y obstrucción basal en el tracto de salida del ventrículo izquierdo (48 mmHg a pesar de 100 mg de atenolol 2 veces al día) e insuficiencia mitral moderada debida a movimiento anterior sistólico de la válvula mitral. La resonancia magnética cardiaca mostró realce tardío de gadolinio (RTG) intramiocárdico en el segmento basal anterior y en el segmento medio inferoseptal (figura 1B-E). En el estudio con Holter de 24 h, se observaron episodios de taquicardia ventricular no sostenida. Se estimó un riesgo de muerte súbita cardiaca del 6,65%1 y se le implantó un desfibrilador automático implantable cuando tenía 25 años. La secuenciación Sanger mostró que este paciente también era portador de p.Ser738Arg.
Las recomendaciones de seguimiento para la MCH incluyen el seguimiento a largo plazo de los portadores sanos de variantes genéticas1. No obstante, no hay aún recomendaciones basadas en variantes específicas. La identificación de variantes relacionadas con un peor pronóstico es clave para tomar decisiones terapéuticas personalizadas.
García-Gustiniani et al.3 informaron de una cohorte de 117 pacientes con MCH portadores de variantes en la región conversora y describieron riesgos altos y bajos que se les asocian.
Se describe una variante en la región conversora del gen MYH7 relacionada con MCH con aparición temprana de hipertrofia miocárdica y fibrosis, y progresión rápida a disfunción sistólica e insuficiencia cardiaca.
Aproximadamente el 3% de los pacientes con MCH contraen disfunción sistólica. La identificación precoz de estos pacientes de alto riesgo es imperativa. Los antecedentes familiares de MCH en estado terminal, diagnóstico a una edad temprana, taquicardia ventricular y presencia y extensión de RTG pueden predecir la aparición de disfunción sistólica en la MCH4. Las mutaciones en la región conversora de MYH7 se han asociado con aparición temprana de MCH y una mayor incidencia de disfunción ventricular izquierda3. La misma región también se ha asociado con un espectro fenotípico que incluye miocardiopatía dilatada, no compactada y restrictiva con algunos sujetos que muestran características solapadas de estos fenotipos3.
Al caso índice en cuestión, se le diagnosticó MCH a una edad temprana con una evolución rápida hacia disfunción sistólica progresiva que requirió trasplante cardiaco. A su hijo se le diagnosticó la miocardiopatía a los 22 años y se le documentó alto riesgo de muerte súbita, por lo que se le implantó un desfibrilador automático implantable cuando tenía 25 años. Este paciente presenta además varios factores de riesgo de disfunción sistólica precoz.
En conclusión, aunque las pruebas genéticas son una herramienta prometedora para evaluar el pronóstico en la MCH, aún están infrautilizadas. La mutación c.2212A > C; p.Ser738Arg en la región conversora del gen MYH7 puede ser un marcador de progresión rápida hacia la fase terminal en la MCH y también podría estar asociada con un mayor riesgo de muerte súbita. Se requieren más estudios y descripciones de casos para aumentar los conocimientos en este campo y poder ofrecer un seguimiento personalizado a los pacientes.