
Se presenta una familia con historia de arritmias auriculares y necesidad de marcapasos (MCP) en varios miembros por enfermedad del nódulo sinusal (ENS) y parálisis auricular. El estudio genético documentó la variante SCN5Ap.Arg219His ya asociada a parálisis auricular familiar (PAF), trastornos de conducción y miocardiopatía dilatada. Hasta donde se sabe, se trata de la primera familia española con PAF descrita.
El caso índice (III.3) es un varón de 30 años sin antecedentes médicos que ingresó por mareo; a su llegada al hospital, se observó en el electrocardiograma (ECG) ausencia de actividad auricular y ritmo idioventricular (figura 1A). El ecocardiograma mostró una aurícula izquierda gravemente dilatada (60 ml/m2) y las demás cavidades tenían dimensiones normales; la resonancia magnética mostró una fracción de eyección del ventrículo izquierdo (FEVI) del 48% y ausencia de realce tardío. Se implantó un MCP bicameral. El paciente evolucionó sin complicaciones y recuperó completamente la FEVI tras 9 meses de tratamiento con ramipril.
. B: electrocardiograma del caso II.3 (62 años). C: electrocardiograma del caso III.8 (32 años). D: electrocardiograma de la portadora asintomática II.8 (58 años).")
Electrocardiogramas de varios portadores de la mutación p.Arg219His en SCN5A. A: electrocardiograma del caso índice III.3 (a los 30 años). B: electrocardiograma del caso II.3 (62 años). C: electrocardiograma del caso III.8 (32 años). D: electrocardiograma de la portadora asintomática II.8 (58 años).
El padre del paciente (II.3) precisó implante de MCP a los 31 años, igualmente por parálisis auricular. En el último ECG alternaba ritmo propio con estimulación VVI (figura 1B). Una tía paterna (II.2) precisó implante de MCP a los 62 años y su hijo (III.1) tuvo un ictus a los 38 años y precisó implante de MCP por ENS. Un primo hermano (III.8) presentaba fibrilación y aleteo auricular desde los 29 años, con varias cardioversiones y ablación de istmo cavotricuspídeo, y en aquel momento se hallaba en ritmo sinusal con propafenona (figura 1C). El último ecocardiograma muestra dilatación biauricular (aurícula izquierda, 45,5 ml/m2) con función biventricular normal.
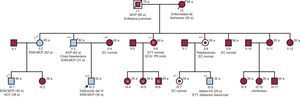
La agregación familiar de ENS con necesidad de MCP a edades tempranas hizo sospechar la existencia de una enfermedad genética con herencia autosómica dominante (figura 2). Se analizaron en el caso índice, mediante ultrasecuenciación, 132 genes relacionados o potencialmente relacionados con arritmias y miocardiopatías. Se encontraron dos mutaciones no presentes en controles en heterocigosis de dos genes previamente relacionados con trastornos de conducción: NC_000003.11:g.38655281C>T en SCN5A y NC_000015.9:g.73615231C>T en HCN4, así como el polimorfismo NC_000021.8:g.35821680C>T en KCNE1 relacionado con intervalo QT largo adquirido.
/HCN4p.Arg1068His (variante de significado incierto).")
Árbol familiar y estudio de las mutaciones SCN5Ap.Arg219His y HCN4p.Arg1068His. Los cuadrados y círculos representan a varones y mujeres respectivamente. Los símbolos sombreados indican individuos afectados. Un punto en el interior del símbolo indica portador no afectado. La letra N en el interior del símbolo indica individuo no portador y no afectado. Los símbolos + y – indican respectivamente individuos testados genéticamente portadores y no portadores de las mutaciones. Una línea diagonal que atraviesa el símbolo indica sujeto fallecido. La edad de un evento aparece entre paréntesis. La edad actual o del fallecimiento aparece en la parte superior derecha del símbolo. a: años; ACV: accidente cerebrovascular; EC: evaluación clínica; ECG: electrocardiograma; ENS: enfermedad del nódulo sinusal; ETT: ecocardiograma transtorácico; FA: fibrilación auricular; MCP: marcapasos; VI: ventrículo izquierdo. SCN5Ap.Arg219His (patogénica)/HCN4p.Arg1068His (variante de significado incierto).
La mutación SCN5Ap.Arg219His se ha descrito en dos familias de Suiza y Japón1,2. Clínicamente, además de ENS, puede cursar con taquiarritmias supraventriculares y ventriculares y necesidad de implante de MCP a edades tempranas, especialmente en varones. En una de las familias se demostró cosegregación de la mutación con miocardiopatía dilatada1,2.
En cambio, la variante HCN4p.Arg1068His no se había descrito previamente. El gen HCN4 codifica la subunidad 4 de los canales HCN, encargados de la corriente nativa If. Participan en la generación y la modulación de la actividad de MCP del corazón. Ciertas mutaciones en HCN4 se asocian a un tiempo de desactivación del canal más rápido que origina pérdida de función y bradicardia3. El estudio in silico de HCN4p.Arg1068His no fue concluyente.
Se ofreció estudio clínico y genético a los familiares previa firma de consentimiento informado. Se realizó estudio con ECG y ecocardiograma a 10 familiares y genético (cuando estaba indicado) a 7.
La mutación SCN5Ap.Arg219His estaba presente en todos los casos afectados de la familia paterna. El fenotipo era muy parecido al descrito previamente, aunque en la literatura la ENS se reporta en casos no emparentados, por lo que es la primera vez que se describe cosegregación de SCN5Ap.Arg219His para PAF1,2 (figura 2). Además se encontró a dos mujeres (II.8 y III.7) portadoras asintomáticas de 58 y 34 años respectivamente, hallazgo que encaja con la presentación clínica más tardía en las mujeres (figura 1D).
La mutación HCN4p.Arg1068His y el polimorfismo KCNE1p.Asp85Asn provenían de la madre, cuyo estudio clínico era normal. No fue posible estudiar a otros miembros de la rama materna. Al no cosegregar con la ENS, se considera que HCN4p.Arg1068His es una variante de significado incierto que no explica la PAF.
La ENS se define como la alteración en la formación y propagación del impulso eléctrico en el nódulo sinusal. Se caracteriza por bradicardia sinusal, bloqueo senoauricular, parada sinusal, incompetencia cronotrópica y/o taquiarritmias auriculares (fundamentalmente fibrilación auricular). Es un trastorno frecuente a edades avanzadas, pero es excepcional en jóvenes. En aproximadamente un 50% de los casos, es necesario implantar un MCP permanente4. Según datos del Registro Español de Marcapasos5, la ENS y la fibrilación auricular/aleteo auricular con bradicardia suponen aproximadamente el 36% de los implantes de MCP. No parece que haya predominancia de sexo (razón varones:mujeres, 0,98) aunque en la fibrilación auricular/aleteo auricular con bradicardia hay predominio de varones (razón, 1,7)5.
Se considera que la mutación SCN5Ap.Arg219His es la causa del cuadro familiar, pues muestra cosegregación con la enfermedad y se ha descrito previamente. Asimismo, la mutación HCN4p.Arg1068His es una variante de significado incierto que podría actuar como modificador de la enfermedad, aunque no la produce. Estos hallazgos permitieron dar consejo genético a los individuos portadores (figura 2). En el caso de los asintomáticos, se ha planificado seguimientos con electrocardiograma, Holter, ecocardiograma (por la asociación con miocardiopatía dilatada) y, según los síntomas, se considerará la realización de ergometría para detectar incompetencia cronotrópica. El estudio familiar, en este caso, fue clave para esclarecer cuál de las variantes genéticas documentadas era la causa de la PAF.
FINANCIACIÓNEste trabajo se ha realizado parcialmente gracias a la financiación del Instituto de Salud Carlos III (PI14/0967, RD12/0042/0049 y RD12/0042/0066).