To the Editor,
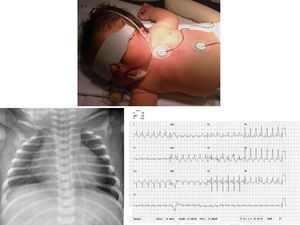
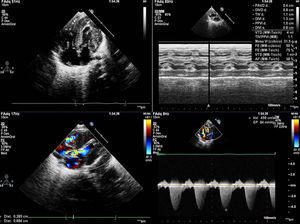
We present the case of a girl diagnosed with prenatal hypertrophic cardiomyopathy in week 32 of gestation. In this first pregnancy for an older woman, the fetus had a thickened nuchal fold at the 12-week screening; amniocentesis revealed a normal 46, XX karyotype and polyhydramnios. She was born at 38 weeks of gestation with no complications. She was admitted for study, and physical examination revealed dysmorphic and somewhat coarse features, macrocephaly and macrosomia, wide chest and short limbs, low-set ears, wide nasal bridge, hypertelorism, antimongoloid slanting eyes, carp mouth with a marked philtrum and high arched palate, low rear hairline, and a short, wide neck (Figure 1); she also had a variable ejection systolic heart murmur in the first few hours of life. The electrocardiogram (Figure 1) showed biventricular hypertrophy with repolarization changes. A chest X-ray (Figure 1) showed moderate cardiomegaly, and an echocardiogram (Figure 2) confirmed the diagnosis of hypertrophic cardiomyopathy with severe obstruction in the left ventricular outflow tract. Treatment was begun with propranolol, and the patient has remained free of cardiac symptoms or arrhythmias. Screening tests for metabolic diseases were normal, and a molecular genetic test for Noonan's syndrome confirmed a KRAS gene mutation. The family study showed the parents were negative.

Figure 1. Phenotype described. X-ray cardiomegaly. Electrocardiographic alterations.

Figure 2. Echocardiogram: ventricular hypertrophy. Severe subaortic obstruction.
Noonan's syndrome is associated with a wide variety of signs and symptoms due to multi-organ involvement. It has some very characteristic signs, such as short stature, while others, although less striking, also can lead to suspicion of the diagnosis. These include a short, wide neck; ears set low and towards the rear, hypertelorism with antimongoloid slanting eyes, epicanthus, ptosis, retromicrognathia and a wide philtrum. There can also be chest and back deformities, joint laxity, chryptorchidism, delayed puberty, coagulation problems, etc. Most patients have normal intelligence, although 15% to 35% have slight retardation, and language problems are common.1
Mutations have been described in 4 genes: PTPN11 (50%), SOS1 (20%), RAF1 (15%) and KRAS (5%); between 10% and 15% are idiopathic.2 In the 35 cases of KRAS mutation, phenotype was more severe than in the other genes.3
Heart defects are very common in this syndrome and their severity varies. The most common are stenotic pulmonary valves (60%) and hypertrophic cardiomyopathies (HCM) (20%). HCM is a primary disease of the heart muscle characterized by hypertrophy of one (generally left) or both ventricles with no cardiac or systemic disease to justify it. Although most cases are idiopathic, several disorders must be ruled out in newborns, the most common being diabetic fetopathies. It may be associated with polymalformation syndromes such as Noonan,4 LEOPARD, Beckwith-Weidemann, or Barth syndromes, and with metabolic diseases, and it is necessary to rule out deposit diseases (glycogenosis, mucopolysaccharidosis, oligosaccharidosis, sphingolipidosis, etc.), energy metabolism disorders (fatty acid oxidative deficiency and carnitine deficiency) and neuromuscular syndromes (Friedrich ataxia5).
The age of presentation of HCM varies greatly and depends above all on the causal mutation, although in many cases this is part of the disease's natural evolution; in older patients, it develops with varying intensities.6 Echocardiography provides a definitive diagnosis, and electrocardiogram monitoring is necessary for the early detection of ventricular arrhythmias, which are sometimes precursors of episodes of sudden cardiac death (SCD), the main cause of mortality. The earlier the age of presentation, the higher the risk of SCD, as these patients also have worse treatment options.7
Beginning pharmacological treatment is recommended in asymptomatic children with obstructive SCD and significant left ventricular outflow tract gradients, and an appropriate stratification of the risk of SCD must be carried out and genetic advice given to parents.8
Corresponding author: tonisanchan@hotmail.com