La enfermedad de Fabry es un trastorno infrecuente y progresivo ligado al cromosoma X, a causa de una deficiencia de la enzima alfagalactosidasa A (α-galA) originada por mutaciones en el gen GLA. La enfermedad motiva una acumulación intracelular de globotriaosilceramida en varios tejidos que afecta a múltiples órganos. En los pacientes varones que padecen la forma clásica de la enfermedad, se observan signos y síntomas precoces en la infancia o la adolescencia1. Esta forma clásica suele aparecer en varones portadores de variantes genéticas que causan una disminución extrema (o ausencia completa) de la actividad enzimática de la α-galA, tal como ocurre con las variantes sin sentido o las que desplazan el marco de lectura2. Las mujeres son heterocigóticas para las mutaciones en el gen GLA y muestran un espectro clínico heterogéneo que oscila entre la ausencia de síntomas y una gravedad clínica igual a la de los varones3.
Se presenta el caso de un paciente con enfermedad de Fabry con un fenotipo relativamente leve pese a ser portador de una mutación sin sentido en GLA relacionada con la variante clásica de la enfermedad.
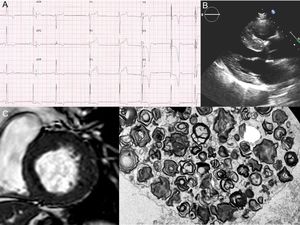
Un varón de 58 años con hipertensión y proteinuria ingresó en el hospital por dolor abdominal. En el electrocardiograma, se observó un intervalo PR corto con hipertrofia del ventrículo izquierdo e isquemia subepicárdica (figura 1A). Se llevó a cabo una angiografía, que mostró una oclusión distal de la segunda rama diagonal; los síntomas mejoraron con el tratamiento médico. El ecocardiograma y la resonancia magnética cardiaca demostraron una hipertrofia septal basal de 20 mm (figura 1B y 1C). Con el diagnóstico de miocardiopatía hipertrófica, se remitió al paciente a la unidad de cardiopatías familiares. Se sospechó enfermedad de Fabry por la miocardiopatía hipertrófica, la proteinuria con leve insuficiencia renal (creatinina, 1,3 mg/dl) y un intervalo PR corto en el electrocardiograma. La actividad de la α-galA en sangre estaba reducida a 0,7 μmol/l/h (2,0-11,7), y la biopsia renal mostró los característicos «cuerpos cebra» en las imágenes de microscopio electrónico (figura 1D). Por último, se secuenciaron 17 genes relacionados con la miocardiopatía hipertrófica, incluido GLA, mediante ultrasecuenciación masiva (next-generation sequencing). Se identificó una ya descrita mutación de truncamiento en GLA (p.Gln386*/g.10021C>T) asociada con las formas clásicas de la enfermedad de Fabry4. En consecuencia, se confirmó el diagnóstico y se inició terapia de reemplazo enzimático.
.")
A: electrocardiograma del paciente que muestra un intervalo PR corto, hipertrofia del ventrículo izquierdo e isquemia subepicárdica. B y C: ecocardiograma y resonancia magnética cardiaca con hipertrofia del ventrículo izquierdo. D: imágenes de microscopio electrónico de la biopsia renal con «cuerpos cebra» (agrupación de cuerpos membranosos y concéntricos de glucolípidos secuestrados dentro de lisosomas).
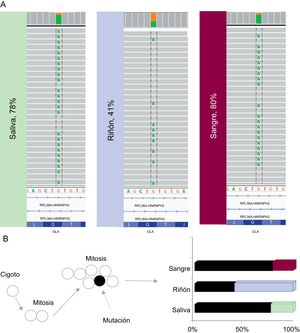
El estudio genético del caso índice se llevó a cabo primero en una muestra de saliva, y en el 78% de las lecturas el alelo estaba mutado, cuando en los portadores hemicigóticos se espera el 100%; la mutación se confirmó mediante secuenciación de Sanger. Se realizaron otros 2 estudios de ultrasecuenciación masiva, en sangre y en muestra renal en parafina (biopsia renal), que mostraron que el alelo estaba mutado en el 80 y el 41% de las lecturas, respectivamente, lo que confirmaba un mosaicismo somático (figura 2A). El método de Sanger es impreciso cuando se trata de cuantificar el porcentaje de mosaicismo y evaluar las diferencias entre los tejidos, pero no la ultrasecuenciación masiva.
 que muestra la mutación (flecha discontinua) y otra (blanca) que muestra el tipo salvaje (wild-type), y diagrama de barras que muestra las lecturas de proporción con el alelo mutado. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
A: las lecturas obtenidas en el estudio de secuenciación de segunda generación de muestras de saliva, riñón y sangre se muestran entre líneas discontinuas negras. B: dos líneas celulares en las células somáticas del paciente, una (negra) que muestra la mutación (flecha discontinua) y otra (blanca) que muestra el tipo salvaje (wild-type), y diagrama de barras que muestra las lecturas de proporción con el alelo mutado. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
La hija y una hermana del probando (de 30 y 55 años respectivamente) que no presentaban ninguna afección no eran portadoras de la variante.
Este paciente presenta un fenotipo que es más leve de lo esperado, tratándose de un varón portador de una mutación de truncamiento, en la que la afección de distintos órganos empieza a una edad temprana y el fenotipo es extremo debido a la total ausencia de la enzima. En este caso en particular, esto puede explicarse por la presencia de 2 líneas celulares en las células somáticas del paciente: una que muestra la mutación y otra wild-type. Así, en cada tejido, hay una proporción de células que pueden mantener cierto grado de actividad enzimática. Esto también se refleja en la actividad de la α-galA de este paciente, que está reducida pero no tanto como se espera en un portador hemicigótico de una mutación de truncamiento en GLA (figura 2B).
La enfermedad de Fabry es hereditaria y se transmite ligada al cromosoma X, por lo que un varón afectado transmitiría la variante patógena a todas sus hijas. En este caso, la descendencia femenina heredaría la mutación en función de la presencia o ausencia del mosaicismo en las células germinales del paciente. En el primer caso (mosaicismo germinal) el riesgo de que las mujeres hereden la mutación se relaciona con la proporción de esperma mutado5. Lamentablemente, esto no se pudo determinar en este paciente porque no se realizaron pruebas genéticas con el tejido espermático y la única hija genotipificada no es portadora de la variante. Hasta la fecha, es el primer caso de enfermedad de Fabry causado por un mosaicismo somático en el gen GLA. El paciente mostró un fenotipo más leve que el esperado para un portador hemicigótico.
Este caso muestra la importancia de las tecnologías de ultrasecuenciación masiva (next-generation sequencing) en los pacientes con sospecha de enfermedad de Fabry, lo que permite un diagnóstico precoz (incluso en caso de mosaicismo somático, como este) y la rápida identificación de aquellos que pueden beneficiarse de la terapia de reemplazo enzimático6.
FINANCIACIÓNFinanciado por una subvención del Centro de Investigación Biomédica en Red (CIBERCV), Instituto de Salud Carlos III, CB16/11/00425 (R. Barriales-Villa y L. Monserrat), Instituto de Salud Carlos III’, FEDER «Unión Europea, Una forma de hacer Europa».
CONFLICTO DE INTERESESJ.P. Ochoa y J.L. Santomé-Collazo son empleados de Health in Code S.L. R. Barriales-Villa ha recibido retribuciones personales de Health in Code S.L. L. Monserrat es parte interesada y CEO de Health in Code S.L.