




Con el término cardiopatías familiares se designa un grupo de enfermedades cardiovasculares (miocardiopatías, canalopatías, algunas enfermedades aórticas y otros síndromes) que comparten una serie de características comunes: tienen una base genética, una presentación familiar, un curso clínico heterogéneo y, por último, todas pueden relacionarse con la muerte súbita. El presente documento recoge de forma resumida algunos conceptos importantes en relación con los avances recientes en las técnicas de secuenciación y el conocimiento de las bases genéticas de estas enfermedades. Se proponen algoritmos diagnósticos y recomendaciones prácticas y se debaten aspectos de interés clínico controvertidos y actuales. Se resalta el papel de las unidades de referencia multidisciplinares para diagnosticarlas y tratarlas.
Palabras clave
Con el término cardiopatías familiares (CF) se designa un grupo de enfermedades cardiovasculares (miocardiopatías, canalopatías, algunas enfermedades aórticas y otros síndromes) que comparten una serie de características comunes:
- 1.
Tienen una presentación familiar. Cuando una persona está afectada, se debe estudiar a los demás miembros de la familia, ya que existe la posibilidad de que también hayan heredado la enfermedad. «Cuando se está ante un paciente con una cardiopatía familiar, no se está evaluando a un paciente, se está estudiando una familia»1. Se estudian familias y, por lo tanto, se debe destacar la coordinación que debe haber con las unidades de cardiología pediátrica.
- 2.
Tienen una base genética. Esto implica que hoy en día se puede hacer un diagnóstico genético de las mismas. Si bien no se logra identificar la mutación causal del 100% de las CF, el porcentaje de mutaciones identificadas ha ido aumentando en los últimos años hasta llegar a cifras superiores al 50% para algunas miocardiopatías y canalopatías, como la miocardiopatía hipertrófica (MCH), la arritmogénica y el síndrome de QT largo (SQTL) (tabla 1).
Tabla 1.Porcentaje de pacientes en los que es posible detectar una mutación causal en función de la cardiopatía familiar
Cardiopatía familiar Estudios genéticos positivos* (%) Miocardiopatía hipertrófica 40-70 (Elliott et al2,3) Miocardiopatía dilatada 30 (Elliott et al3, Ackerman et al4) Miocardiopatía restrictiva Desconocido (Elliott et al3, Ackerman et al4) Miocardiopatía no compactada 17-41 (Elliott et al3, Ackerman et al4) Miocardiopatía (displasia) arritmogénica 60 (Elliott et al3, Ackerman et al4) Síndrome de Brugada 20-30 (Ackerman et al4) TVCP 60-70 (Ackerman et al4) Síndrome de QT largo 70-80 (Ackerman et al4) Síndrome de QT corto Desconocido (Ackerman et al4) Síndrome de Marfan 70-93 (Loeys et al5) Síndrome de Loeys-Dietz Depende de la evaluación clínica/imagen (Arslan-Kirchner et al6) TVCP: taquicardia ventricular catecolaminérgica polimórfica.
- 3.
Pueden ser causa de muerte súbita, en ocasiones como forma de presentación de la enfermedad. La muerte súbita tiene un alto impacto social, económico y mediático. La cardiopatía isquémica es la causa más importante de muerte súbita en individuos de edad avanzada con factores de riesgo de enfermedad coronaria, mientras que las CF son causa frecuente en menores de 35 años (ya sean deportistas o no)7,8.
En los últimos años se han publicado a nivel internacional diferentes documentos de consenso y guías clínicas sobre diagnóstico y tratamiento de las miocardiopatías, canalopatías y diversas enfermedades aórticas de origen genético. En todos ellos se incide en la necesidad (con diferentes niveles de recomendación y evidencia) de estudiar a los familiares de los pacientes afectados, así como en la realización de estudios genéticos (tabla 2).
Recomendaciones y nivel de evidencia de los estudios genéticos en las guías y los documentos de consenso publicados
| Estudio genético | Nivel de recomendacióna | Nivel de evidencia |
|---|---|---|
| Miocardiopatía hipertrófica (Elliott et al2, Ackerman et al4) | I | B |
| I | C | |
| Miocardiopatía dilatada (Ackerman et al4) | Ib | C |
| IIac | ||
| Miocardiopatía restrictiva (Ackerman et al4) | IIb | C |
| Miocardiopatía no compactada (Ackerman et al4) | IIa | C |
| Miocardiopatía arritmogénica (Ackerman et al4) | IIa (incluido en los criterios diagnósticos) | C |
| Síndrome de Brugada (Ackerman et al4) | IIa | C |
| Taquicardia ventricular polimórfica catecolaminérgica (Ackerman et al4) | I (incluido en los criterios diagnósticos) | C |
| Síndrome de QT largo (Ackerman et al4) | I (incluido en los criterios diagnósticos) | C |
| Síndrome de QT corto (Ackerman et al4) | IIb (incluido en los criterios diagnósticos) | C |
| Síndrome de Marfan (Loeys et al5) | Incluido en los criterios diagnósticos | |
| Síndrome de Loeys-Dietz (Arslan-Kirchner et al6) | Incluido en los criterios diagnósticos |
I: se recomienda; IIa: puede ser útil; IIb: se puede considerar.
También se hace mención a la creación de unidades especializadas, de referencia, para el diagnóstico y el tratamiento de estas CF. Estas unidades deben ser multidisciplinarias y mantener una estrecha relación con otros servicios y especialidades2,9. Sin embargo, estas unidades de referencia no existen en todas las comunidades de España, por lo que en ocasiones se hace difícil el cumplimiento de muchas de las recomendaciones de estos documentos.
El presente documento surge desde el Grupo de Trabajo de Cardiopatías Familiares de la Sociedad Española de Cardiología (SEC) y cuenta con la colaboración del Grupo de Trabajo de Aorta (de la Sección de Cardiología Clínica), el Grupo de Trabajo de Cardiología del Deporte, la Sección de Electrofisiología y Arritmias, la Sección de Insuficiencia Cardiaca y Trasplante Cardiaco de la SEC y la Sociedad Española de Cardiología Pediátrica y Cardiopatías Congénitas. Además, se cuenta con un grupo de expertos nacionales e internacionales que han revisado el documento (anexo).
Se ha creído conveniente explicar diversos aspectos de la atención a los pacientes con CF, con el fin de acercar las diferentes guías y recomendaciones a la práctica clínica española.
Actualmente existen en España varias unidades de referencia designadas por el Ministerio de Sanidad (Centros, Servicios y Unidades de Referencia en el Sistema Nacional de Salud) para el diagnóstico y el tratamiento de las CF10. Las unidades de referencia son muy necesarias para casos complejos, de difícil diagnóstico y/o tratamiento, pero es muy importante exportar la «manera de trabajar» de las unidades de CF a todos los profesionales sanitarios (cardiólogos, internistas, cirujanos cardiovasculares, genetistas, pediatras, enfermeras, psicólogos, etc.) que puedan verse implicados en el estudio y el tratamiento de estas enfermedades.
Además, se ha creído adecuado detenerse en determinados aspectos y terminologías que merecen una atención especial debido a su novedad o sus características multidisciplinarias. Por último, se destacan determinadas recomendaciones (tabla 3), consideradas básicas y que hay que tener en cuenta como punto de partida para avanzar en el conocimiento y el tratamiento de estas enfermedades.
Recomendaciones para el manejo de las cardiopatías familiares
| I | Se debe dibujar un árbol familiar de al menos tres generaciones, preguntando una a una por posibles enfermedades relacionadas con la CF en estudio. El esfuerzo por recordar síntomas y signos de los parientes no solo permite detectar en ocasiones a otros miembros afectados o posiblemente afectados, sino que también facilita el reconocimiento del patrón de herencia |
| II | Ante un paciente con una CF, es necesario evaluar a sus familiares. La evaluación de los diferentes miembros de una familia debe realizarla un especialista o una unidad con experiencia integrando los resultados de todos sus miembros. Se debe ofrecer seguimiento a los familiares en riesgo de presentar la enfermedad. El estudio genético puede facilitar el estudio familiar en caso de que se haya identificado una mutación patogénica en la familia |
| III | Los estudios genéticos deben incluirse en el arsenal diagnóstico clínico habitual en las CF. Las indicaciones para realizarlo dependen de la rentabilidad en la patología (porcentaje de positivos) y el valor de su resultado en el diagnóstico y el pronóstico de los afectados y sus familiares |
| IV | En la atención a pacientes con CF, se debe excluir específicamente una cardiopatía causada por fenocopia, ya que estas entidades tienen habitualmente un curso clínico y un tratamiento diferenciados |
| V | Es recomendable comenzar el estudio clínico de los familiares de pacientes con CF independientemente de la edad. En cuanto al estudio genético de los familiares de pacientes con miocardiopatías o enfermedades aórticas hereditarias, se recomienda a los 10 años de edad (en el momento del diagnóstico en el caso de las canalopatías). Esta edad es orientativa y se debe modificar según la gravedad del fenotipo familiar y las particularidades de cada familia y cada centro de estudio |
| VI | Se debe dar asesoramiento reproductivo a los pacientes que deseen tener descendencia. El asesoramiento deben brindarlo unidades expertas en la patología, la legislación y las técnicas de reproducción. La legislación española debería realizar un esfuerzo para adaptarse a la nueva realidad de las CF y en este documento se propone la creación de grupos de trabajo para avanzar en este campo y facilitar la tarea |
| VII | Se debe recoger y almacenar muestras de sangre y/o tejido de los fallecidos súbitamente con CF sospechada o confirmada que permitan la realización de un estudio genético («autopsia molecular»). Sería deseable que en cada comunidad autónoma se facilitaran los medios para la creación de centros de referencia para el estudio macroscópico y microscópico de los corazones y las aortas de los pacientes jóvenes con sospecha de CF fallecidos súbitamente. Estas unidades deberían trabajar de manera coordinada con las unidades de CF. Es recomendable que a su vez se establezcan lazos de colaboración estables entre las administraciones de justicia (de las que dependen forenses y patólogos forenses) y sanidad (de la que dependen las unidades de CF) |
| VIII | Sería deseable la incorporación de psicólogos o personal especializado en atención psicológica en las unidades de CF |
| IX | Se debe fomentar y ayudar a los pacientes con CF en la creación de asociaciones de pacientes que den apoyo adicional no solo a los afectados, sino también a los familiares sanos que conviven con ellos |
CF: cardiopatía familar.
Un árbol familiar, árbol genealógico o pedigree médico es una representación gráfica del historial médico y los parentescos de una familia. Con los recientes avances de la genética y su aplicación cada vez más extendida, cualquier médico (o profesional de la salud) debería poder dibujar e interpretar un árbol familiar.
Se recomienda la realización de un árbol familiar detallado, de tres generaciones, como parte de la evaluación de un paciente con una CF (figura 1). Las preguntas deben estar dirigidas a extraer la información relevante relacionada con los antecedentes cardiológicos y síntomas que indiquen un origen cardiaco11,12.

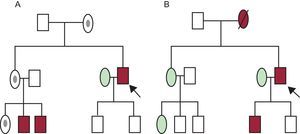
Árboles familiares o pedigrees de dos familias con una cardiopatía familiar. A: transmisión ligada al cromosoma X. B: transmisión autosómica dominante. Rectángulo: varón; óvalo: mujer; rectángulo u óvalo en granate: varón o mujer fenotípicamente afectados; rectángulo u óvalo sin relleno: varón o mujer sanos; punto negro en interior de óvalo: portadora sana; flecha: probando o caso índice; línea diagonal que tacha un símbolo: persona fallecida. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Así, es importante: a) identificar al sujeto índice (o probando), que es el primer caso valorado en la familia (puede estar vivo o haber fallecido); b) anotar nombres, fechas de nacimiento y las causas y edades de los fallecimientos (se incluyen los casos de muerte súbita del lactante), siempre respetando las leyes de protección de datos vigentes; c) es importante descartar la existencia de consanguinidad y preguntar por los orígenes geográficos de la familia; d) indagar sobre síntomas, signos, complicaciones, tratamientos (desfibriladores, marcapasos, trasplante cardiaco/renal, etc.) que puedan relacionarse con la CF en estudio, y e) la investigación familiar incluye una revisión de toda la documentación disponible (historias clínicas, electrocardiogramas antiguos, etc.). En este sentido es fundamental localizar los informes de autopsias, en su caso.
TIPOS DE HERENCIALa mayoría de las CF son de transmisión autosómica dominante, es decir, el paciente puede transmitir la enfermedad tanto a varones como a mujeres y sus descendientes tienen un 50% de probabilidades de heredar el defecto genético que causa la enfermedad. Si se observa una «transmisión de la enfermedad de varón a varón» dentro del árbol familiar, se confirma la herencia autosómica dominante.
Cuando hay antecedentes de consanguinidad, es más probable la herencia autosómica recesiva, y en ese contexto cada descendiente tiene un 25% de posibilidades de heredar los dos alelos mutados. Una transmisión de mujer a varón con la madre sana y el varón afectado es altamente indicativa de enfermedad recesiva ligada al cromosoma X. Una transmisión de mujer a mujer o varón sin transmisión del varón a la descendencia indica herencia matrilineal (típica de las enfermedades mitocondriales). En ocasiones, la mutación puede aparecer de novo en el caso índice, de modo que no la tienen los padres de este, que sí puede transmitirla a los descendientes11,13.
Recomendación I: se debe dibujar un árbol familiar de al menos tres generaciones, preguntando una a una por posibles enfermedades relacionadas con la CF en estudio. El esfuerzo de recordar síntomas y signos de los parientes no solo permite detectar en ocasiones a miembros afectados o posiblemente afectados, sino que también facilita el reconocimiento del patrón de herencia.
CRITERIOS DIAGNÓSTICOS: PARA CASOS ÍNDICES Y PARA FAMILIARESEn la tabla 4 se recoge los documentos de consenso o guías que describen los criterios diagnósticos de las principales CF.
Documentos de consenso o guías en los que se describen los criterios diagnósticos de las principales cardiopatías familiares
| Cardiopatía familiar | Documento |
|---|---|
| Miocardiopatía hipertrófica | 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (Elliott et al2)Comentarios a la guía de práctica clínica de la European Society of Cardiology 2014 sobre el diagnóstico y manejo de la miocardiopatía hipertrófica. Una visión crítica desde la cardiología española (Grupo de Trabajo de la SEC14) |
| Miocardiopatías | Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases (Charron et al11) |
| Canalopatías | HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes (Priori et al9) |
| Miocardiopatía dilatada* | Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy (Mestroni et al15) |
| Miocardiopatías y canalopatías | HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies (Ackerman et al4) |
| Miocardiopatías | Genetic Evaluation of Cardiomyopathy: A Heart Failure Society of America Practice Guideline (Hershberger et al16) |
| Miocardiopatía arritmogénica | Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria (Marcus et al17) |
| Enfermedad de la aorta torácica | 2010 CCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine (Hiratzka et al18) |
| Síndrome de Marfan | The revised Ghent nosology for the Marfan syndrome (Loeys et al5) |
| Enfermedad aórtica | 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (Erbel et al19)Comentarios a la guía de práctica clínica de la ESC 2014 sobre diagnóstico y tratamiento de la patología de la aorta (Grupo de Trabajo de la SEC20) |
AATS: American Association for Thoracic Surgery; ACCF: American College of Cardiology Foundation; ACR: American College of Radiology; AHA: American Heart Association; APHRS: Asia Pacific Heart Rhythm Society; ASA: American Stroke Association; EHRA: European Heart Rhythm Association; ESC: European Society of Cardiology; HRS: Heart Rhythm Society; SCA: Society of Cardiovascular Anesthesiologists; SCAI: Society for Cardiovascular Angiography and Interventions; SEC: Sociedad Española de Cardiología; SIR: Society of Interventional Radiology; STS: Society of Thoracic Surgeons; SVM: Society for Vascular Medicine.
En algunas CF se distinguen criterios que se deben aplicar al caso índice y criterios, en general menos estrictos, que deben aplicarse a los familiares, que en definitiva facilitan el diagnóstico de la enfermedad en estadios más iniciales. El diagnóstico temprano en familiares puede implicar cambios de hábitos de vida e inicio de tratamientos e incidir positivamente en el pronóstico de la enfermedad.
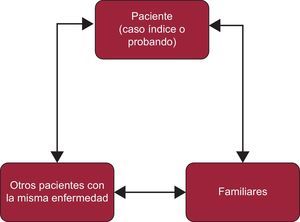
PROTOCOLOS Y ALGORITMOS DIAGNÓSTICOSEn las CF se debe estudiar no solo al caso índice o probando (afectado), sino también a sus familiares (que pueden estar afectados o no). La información que se obtenga del caso índice va a ser vital para el estudio de los familiares. Del mismo modo, la información de los familiares puede ser fundamental para establecer un diagnóstico definitivo en el caso índice (figura 2).

En el diagnóstico y el tratamiento de enfermedades poco frecuentes como las CF, se hacen imprescindibles los registros tanto locales como nacionales o internacionales.
A continuación se proponen algoritmos diagnósticos generales para las dos situaciones más frecuentes en la consulta de CF:
- 1.
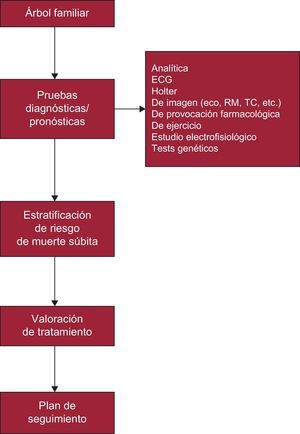
Paciente con sospecha diagnóstica de CF (miocardiopatía, canalopatía, enfermedad aórtica, etc.) (figura 3).
- 2.
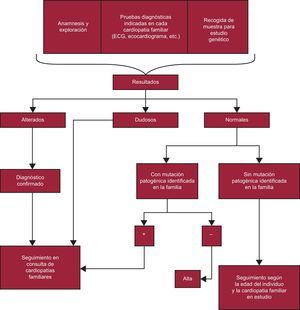
Familiar de paciente con determinada CF (figura 4).
 y por la posibilidad de que en la familia haya más de una mutación (p. ej., podría haber un familiar afectado que sea portador de la mutación no identificada en el estudio genético realizado). Se debería mantener esta actitud con todos los estudios familiares de las diferentes cardiopatías. ECG: electrocardiograma.") Figura 4.
Figura 4.Algoritmo en el que se esquematiza la actuación con los familiares de un paciente diagnosticado de una cardiopatía familiar. Según las recomendaciones de la guía para el diagnóstico y el tratamiento de la miocardiopatía hipertrófica de la Sociedad Europea de Cardiología de 2014, se puede dar el alta al paciente sin haberle hecho una valoración clínica con ecocardiograma y electrocardiograma si no es portador de la mutación familiar. El Grupo de Trabajo de Cardiopatías Familiares de la Sociedad Española de Cardiología opina que no se debe dar el alta a ningún familiar de un paciente con una miocardiopatía sin previamente realizarle una ecocardiografía y un electrocardiograma, por la utilidad de estas exploraciones para establecer la cosegregación de la mutación en cada familia que respalde su patogenicidad (con mutaciones muchas veces de novo, sin experiencia publicada en la bibliografía) y por la posibilidad de que en la familia haya más de una mutación (p. ej., podría haber un familiar afectado que sea portador de la mutación no identificada en el estudio genético realizado). Se debería mantener esta actitud con todos los estudios familiares de las diferentes cardiopatías. ECG: electrocardiograma.
(0.33MB).

 y por la posibilidad de que en la familia haya más de una mutación (p. ej., podría haber un familiar afectado que sea portador de la mutación no identificada en el estudio genético realizado). Se debería mantener esta actitud con todos los estudios familiares de las diferentes cardiopatías. ECG: electrocardiograma.")
Con el fin de completar el diagnóstico y establecer el pronóstico de la familia, el médico responsable de la consulta o la unidad de CF deben integrar toda la información recogida de las dos partes.
Recomendación II: ante un paciente con una CF, es necesario evaluar a sus familiares. Un especialista o una unidad con experiencia deben evaluar a los diferentes miembros de una familia integrando los resultados de todos sus miembros. Se debe ofrecer seguimiento a los familiares en riesgo de presentar la enfermedad. El estudio genético puede facilitar el estudio familiar en caso de que se haya identificado una mutación patogénica en la familia.
GENÉTICALos estudios genéticos han cambiado radicalmente la actitud ante las CF y son una herramienta fundamental en los algoritmos diagnósticos. Hasta hace unos años, el estudio se limitaba a unos pocos genes y con un coste muy elevado (método Sanger). Actualmente, con el desarrollo de técnicas de secuenciación masiva (next-generation sequencing [NGS]), es posible estudiar cientos de genes o incluso todo el genoma (o la parte codificante, conocida como exoma) a precios razonables y rápidamente. El principal problema de la NGS estriba en que, al estudiar tantos genes, se puede detectar múltiples variantes cuya patogenicidad es difícil de evaluar21.
Todas las guías o documentos de consenso recientes sobre las CF ya incluyen los estudios genéticos con los más altos niveles de recomendación (tabla 2).
Los estudios genéticos en las CF tienen utilidad diagnóstica y aplicación directa en cuanto a la posibilidad de ofrecer consejo reproductivo/profesional y organizar el seguimiento de familiares. En algunos casos, el estudio genético tiene también implicaciones en el tratamiento y el pronóstico de los pacientes4,22,23.
Recomendación III: los estudios genéticos deben estar incluidos en el arsenal diagnóstico clínico habitual de las CF. Las indicaciones para realizarlos dependen de la rentabilidad en la patología (porcentaje de positivos) y el valor de su resultado en el diagnóstico y el pronóstico de los afectados y sus familiares.
FENOCOPIASCon el término fenocopia se designa a pacientes que presentan el mismo fenotipo (rasgos característicos de una enfermedad) que otros individuos que tienen una alteración genética conocida que ellos no presentan3. Existen fenocopias tanto de miocardiopatías como de canalopatías y enfermedades aórticas.
Sin embargo, se cree que este concepto es confuso. Por ejemplo, se considera fenocopias de la MCH a las «hipertrofias ventriculares» producidas por mutaciones en el gen GLA (enfermedad de Fabry) o en LAMP2 (enfermedad de Danon), entre otras muchas. Sin embargo, de acuerdo con la clasificación europea de miocardiopatías de 20083, que las agrupa según la morfología y la función ventricular, todas ellas son MCH. El término fenocopia en la MCH se basa en considerar MCH solo a las hipertrofias debidas a mutaciones en genes sarcoméricos. En sentido estricto, se podría emplear el término fenocopia en la hipertrofia debida al «corazón de deportista»24.
Otro ejemplo es la taquicardia ventricular catecolaminérgica polimórfica, que se diagnostica por un corazón estructuralmente normal con electrocardiograma normal y arritmias ventriculares polimórficas en ejercicio o en el test de adrenalina. La taquicardia ventricular catecolaminérgica polimórfica se debe, en su mayor parte, a mutaciones en RYR2 y CASQ2. Sin embargo, mutaciones en otros genes relacionados con distintos tipos de síndrome de QT largo también pueden producir arritmias que recuerdan la descripción clásica de la taquicardia ventricular polimórfica catecolaminérgica, como en el síndrome de Andersen-Tawil (por mutaciones en KCNJ2 y síndrome de QT largo tipo 7) o las mutaciones en ANK2 (síndrome de QT largo tipo 4)4.
También puede ocurrir en el síndrome de Marfan, que se produce por mutaciones FBN1 (gen de la fibrilina 1). Hace unos años se distinguían dos tipos de síndrome de Marfan: el tipo 1 debido a mutaciones en FBN1 y el tipo 2 debido a mutaciones en TGFBR1 y TGFBR2. Posteriormente, se agrupó a estos pacientes con características marfanoides, pero con enfermedad vascular agresiva y presencia de otras características morfológicas como hipertelorismo, úvula bífida, tortuosidad arterial, etc., en el síndrome de Loeys-Dietz13.
Es importante conocer estas «fenocopias», porque algunas pueden tener un comportamiento más agresivo que la enfermedad que «imitan» y porque para muchas de ellas existen tratamientos específicos (tratamiento enzimático sustitutivo en el Fabry) o hay que tomar medidas terapéuticas más agresivas (cirugía más precoz en el síndrome de Loeys-Dietz). Así, los protocolos diagnósticos en las CF deben incluir todas las pruebas de imagen o de laboratorio que descarten una posible fenocopia.
FENOTIPOS SOLAPADOSEntre las miocardiopatías, se puede encontrar los siguientes fenotipos: MCH, restrictiva, dilatada, arritmogénica y no compactada4. Clásicamente se las consideró enfermedades diferentes e independientes, pero hoy se sabe que se puede encontrar a pacientes que evolucionan de un fenotipo a otro (p. ej., paciente con MCH que evoluciona a miocardiopatía dilatada) o que en una misma familia hay varios fenotipos todos ellos asociados a una misma mutación (paciente con miocardiopatía no compactada y familiares con MCH). En las canalopatías también se produce un fenómeno similar, ya que, por ejemplo, se han descrito mutaciones en SCN5A que pueden causar síndrome de QT largo o síndrome de Brugada25.
La genética tiene un papel fundamental en el conocimiento de estas enfermedades. Estos «fenotipos solapados» pueden deberse a que mutaciones en diferentes regiones de un mismo gen pueden tener diferentes efectos, pero también hay que considerar la posibilidad de que, aun con un fenotipo distinto, se esté hablando de la misma enfermedad.
Recomendación IV: en la atención a pacientes con CF, se debe excluir específicamente una cardiopatía causada por una fenocopia, ya que estas entidades habitualmente tienen un curso clínico y un tratamiento diferenciales.
INTERPRETACIÓN DEL ESTUDIO GENÉTICODe manera simplificada, se considera25:
- 1.
Clasificación por frecuencia:
- a.
Polimorfismo: se trata de un cambio en la secuencia de nucleótidos que está presente también en la población general sana (al menos en un 0,5-1,0%).
- b.
Variantes raras: cuando estos cambios aparecen en un porcentaje muy bajo de la población general sana (< 0,5%).
- c.
Mutaciones: cambios que no aparecen en la población sana.
- a.
- 2.
Clasificación por patogenicidad: las enfermedades monogénicas, como la mayoría de las CF, están causadas por mutaciones o variantes raras; la mayoría de los polimorfismos no son patogénicos, pero en algunos casos pueden existir algunos polimorfismos que actúen como moduladores o modificadores que, en presencia de determinados factores ambientales (p. ej., medicamentos), podrían producir enfermedad. Así:
- a.
La presencia de una variante patogénica (habitualmente una mutación) sirve para confirmar el diagnóstico en el caso índice, completar el estudio y el seguimiento de los familiares portadores y, en ocasiones, conocer el pronóstico y dar el alta de la consulta a los familiares no portadores.
- b.
La identificación de una variante benigna (habitualmente un polimorfismo) en el caso índice no sirve para confirmar el diagnóstico ni para el estudio de los familiares, salvo que se haya demostrado su acción como modificador o modulador.
- c.
Variantes de patogenicidad desconocida o de significado incierto (VUS [variants of unknown significance]). Son cambios en la secuencia de nucleótidos de los que actualmente, con el conocimiento disponible, se desconoce si producen enfermedad o no. No se puede utilizar las VUS ni para confirmar el diagnóstico ni para el estudio de los familiares. Se podría estudiar algunas de estas variantes en la familia, pero con fines de investigación. En un reciente documento de consenso, la Sociedad Europea de Cardiología recomienda mantener contacto con los pacientes portadores de VUS, por si en un futuro se establece su patogenicidad11,21.
- a.
Por lo tanto, la realización de un estudio genético en una CF no significa que siempre se detecte la mutación patogénica causante de la enfermedad. El porcentaje de positividad del estudio genético oscila entre el 20 y el 90%, dependiendo de la enfermedad (tabla 1). No encontrar una mutación patogénica en una familia diagnosticada por criterios clínicos de CF no significa que no presente la enfermedad. Dado que no se conoce la totalidad de los genes implicados en todas las CF, se debe considerar afectada a esa familia y se debe evaluar a sus integrantes de manera acorde con los criterios clínicos.
¿A QUÉ EDAD SE RECOMIENDA COMENZAR EL ESTUDIO CLÍNICO Y GENÉTICO DE FAMILIARES DE PACIENTES CON CARDIOPATÍAS FAMILIARES?Este es un aspecto muy controvertido. Los documentos de consenso y guías realizan recomendaciones que, en ocasiones, son poco concretas, lo que deja al médico y la familia la última decisión. La Sociedad Europea de Genética recomienda retrasar el momento de realizar los estudios genéticos de los niños y argumenta que el diagnóstico puede añadir fuertes cargas de ansiedad no justificadas, así como actitudes de sobreprotección26. Por otro lado, un gran número de cardiólogos (incluidos los cardiólogos infantiles) esgrimen que las CF se viven con mayor ansiedad por la incertidumbre del diagnóstico, más que por el conocimiento del estado de portador o afectado.
Considerando que lo más frecuente es que el patrón de herencia sea autosómica dominante, el 50% de la descendencia no heredará el defecto genético y no precisaría revisiones periódicas ni restricciones físicas o farmacológicas (caso de las canalopatías).
Las muertes súbitas a edades tempranas en la familia, el deseo de practicar deporte o iniciarse en el deporte de competición o el deseo irrefrenable de la familia de saber si su hijo tiene la mutación familiar o no pueden justificar un estudio genético antes de lo idealmente recomendado2,4,6,11,13. El debate sobre el momento de realizar los estudios genéticos sigue abierto.
Los autores de este documento consideran que los familiares deben pasar por una evaluación clínica independientemente de la edad, tanto por miocardiopatías como por canalopatías o enfermedades aórticas y, tras la evaluación inicial, deben mantenerse las revisiones periódicas según la enfermedad y la gravedad del fenotipo.
Ahora bien, en cuanto al estudio genético, hay una clara diferenciación entre las enfermedades estructurales (miocardiopatías y enfermedades genéticas de la aorta) y las no estructurales (canalopatías). En las primeras, el riesgo de complicaciones suele ir ligado a la presencia y la gravedad del fenotipo, por lo que, si se decide una actitud conservadora, es posible esperar hasta la mayoría de edad para que el paciente decida si se somete a estudio genético, ya que, en ausencia de expresión fenotípica, la actitud terapéutica va a cambiar poco o nada. De todos modos, se reconoce que una actitud «intervencionista» con diagnóstico genético precoz también es posible siempre que el equipo médico interprete correctamente los resultados y los explique a la familia convenientemente.
Sin embargo, ante las canalopatías, se cree indicada la realización temprana del estudio genético, ya que sus resultados conllevan cambios en hábitos de vida dirigidos a prevenir complicaciones arrítmicas importantes.
Recomendación V: es recomendable comenzar el estudio clínico de los familiares de pacientes con CF independientemente de la edad.
En cuanto al estudio genético de los familiares de pacientes con miocardiopatías o enfermedades aórticas hereditarias, se recomienda a los 10 años de edad (o en el momento del diagnóstico en caso de canalopatías). Esta edad es orientativa y se debe modificar de acuerdo con la gravedad del fenotipo familiar y las particularidades de cada familia y cada centro de estudio.
ASESORAMIENTO REPRODUCTIVOUna de las consecuencias derivadas de la realización de estudios genéticos en las CF es su repercusión sobre la reproducción. El objetivo debe ser que el paciente conozca las posibilidades de trasmitir la alteración genética a la descendencia, las implicaciones de esta trasmisión y qué alternativas reproductivas tiene.
Muchas parejas consultan por la posibilidad de tener descendencia que esté libre de la enfermedad familiar. Existen diferentes posibilidades para evitar la transmisión del defecto genético, pero todas ellas están sujetas a aspectos éticos y legales importantes que se debe tener en cuenta:
- 1.
El diagnóstico prenatal analiza el ADN extraído de células fetales obtenidas de las vellosidades coriónicas (entre las semanas 10 y 12) o de células del líquido amniótico (entre las semanas 14 y 16 de gestación). Podría realizarse si previamente se ha identificado una mutación causal en la familia, siempre que esa mutación tenga una patogenicidad claramente demostrada, se espere discapacidad o haya alto riesgo de muerte precoz y no existan posibilidades de tratamiento eficaces. Dado que las técnicas de diagnóstico prenatal implican riesgo de aborto, solo estarían justificadas si se pretende interrumpir el embarazo en caso de que se demuestre que el feto es portador del defecto genético.
- 2.
El diagnóstico preimplantacional se realiza asociado a técnicas de fecundación in vitro. Permite estudiar si los embriones fecundados in vitro son portadores de la mutación patogénica o no, de modo que solo se implantarán los que no la tengan. La legislación española permite realizar estas técnicas para detectar enfermedades hereditarias graves, de aparición precoz y no susceptibles de tratamiento curativo27. Sin embargo, la ausencia de mención explícita de qué enfermedades pueden beneficiarse de estos procedimientos en esta ley es fuente de confusión. Estas técnicas están plenamente aceptadas para algunas CF como la enfermedad de Marfan o la enfermedad de Fabry. Para las demás CF, es necesario solicitar la autorización individual de la Comisión Nacional de Reproducción Asistida13,27.
- 3.
Otras posibilidades que aseguran la ausencia de trasmisión del defecto genético familiar son el empleo de ovocitos o espermatozoides de donante (dependiendo del progenitor afectado) y la adopción.
Recomendación VI: se debe dar asesoramiento reproductivo a los pacientes que deseen tener descendencia. Deben brindar el asesoramiento unidades expertas en la patología, la legislación y las técnicas de reproducción. La legislación española debería realizar un esfuerzo para adaptarse a la nueva realidad de las CF, y en este documento se propone la creación de grupos de trabajo para avanzar en este campo y facilitar esta tarea.
ESTUDIO DE LA MUERTE SÚBITALa muerte súbita, particularmente cuando ocurre en una persona joven (menor de 40 años), puede deberse a una CF7,8. El diagnóstico correcto de la causa del fallecimiento puede evitar que se repita un desenlace fatal en otros miembros de la familia.
Tras la muerte súbita de una persona joven, debe realizarse una autopsia completa siguiendo protocolos específicos para conocer la causa en profundidad28–30. Esta autopsia debería prestar especial atención al corazón y la aorta, y es recomendable el estudio por forenses y/o patólogos expertos en CF. Incluso en manos expertas, en una porción variable de los casos no se identifica la causa de la muerte (entre el 4 y el 50% de las autopsias medicolegales; los mayores porcentajes corresponden a series forenses de niños y adolescentes30–32).
Es fundamental conservar muestras de tejidos del fallecido que permitan revisar el caso desde el punto de vista patológico con diferentes técnicas o tinciones. Para el estudio molecular o genético, es preciso conservar muestras de sangre o tejido (sin fijar). El estudio genético, lo que se conoce como «autopsia molecular», puede facilitar el diagnóstico de la causa de muerte en más del 35% de los casos en que la autopsia es negativa33.
Se consiga o no un diagnostico definitivo en la autopsia, y tras descartar una enfermedad cardiaca no hereditaria (fundamentalmente isquémica o valvular), se recomienda realizar un estudio cardiológico de los familiares de primer grado (más o menos extenso dependiendo de la enfermedad que se sospecha).
En los documentos de expertos y en las guías9,34 se recomienda específicamente el desarrollo de consultas de CF (o de «arritmias familiares») para el estudio de las causas de muerte súbita.
Recomendación VII: se debe recoger y almacenar muestras de sangre y/o tejido de los fallecidos de muerte súbita con CF sospechada o confirmada que permitan el estudio genético («autopsia molecular»). Sería deseable que en cada comunidad autónoma se facilitaran los medios para la creación de centros de referencia para el estudio macroscópico y microscópico de los corazones y las aortas de los pacientes jóvenes con sospecha de CF fallecidos súbitamente. Estas unidades deberían trabajar de manera coordinada con las unidades de CF. Es recomendable que a su vez se establezcan lazos de colaboración estables entre las administraciones de justicia (de las que dependen forenses y patólogos forenses) y sanidad (de la que dependen las unidades de CF).
APOYO AL PACIENTE Y A LAS FAMILIASAunque una gran mayoría de los pacientes afectados de una CF pueden llevar una vida normal, un pequeño porcentaje presenta discapacidades importantes o muertes prematuras. Afrontar el fallecimiento repentino de un familiar, particularmente cuando se trata de un niño o un joven, no es tarea fácil.
En España salvo excepciones, la sanidad pública no cuenta con unidades de psicología especializadas para estas situaciones. En otras áreas de la medicina, como la oncología, sí hay larga experiencia en programas de apoyo psicológico.
En otros países del entorno, como Reino Unido35,36, existen asociaciones de pacientes que desempeñan una labor encomiable en el apoyo, el seguimiento, la formación (p. ej., en técnicas de reanimación cardiopulmonar básica) y la información de estos pacientes. En España, aunque existen algunas asociaciones37,38, no son suficientes y se echa en falta este elemento asociativo que tantos beneficios puede aportar al paciente. La información que los médicos pueden dar a los pacientes es importante, pero nadie como un paciente que tiene la misma enfermedad para explicar a otro paciente cómo son las sensaciones y vivencias de esta.
Recomendación VIII: sería deseable la incorporación de psicólogos o personal especializado en atención psicológica en las unidades de CF.
Recomendación IX: se debe fomentar y ayudar a los pacientes con CF en la creación de asociaciones de pacientes que den apoyo adicional no solo a los afectados, sino también a los familiares sanos que conviven con ellos.
CONCLUSIONESCon este documento se trata de exportar la «manera de trabajar» de las unidades de CF a todos los profesionales sanitarios (cardiólogos, internistas, cirujanos cardiovasculares, genetistas, pediatras, enfermeras, psicólogos, etc.) que puedan verse implicados en el estudio y el tratamiento de estas enfermedades. Se proponen algoritmos diagnósticos, se debaten aspectos actuales de interés clínico controvertidos y se destacan nueve recomendaciones básicas que tener en cuenta como punto de partida para avanzar en el conocimiento y el tratamiento de estas enfermedades. Además de su indudable valor como documento de síntesis del estado actual del manejo de las CF, este documento puede ayudar a homogenizar el proceso asistencial de estos pacientes en España, lo que sin duda redundará en una mejora de la calidad asistencial.
FINANCIACIÓNEste trabajo ha sido parcialmente financiado por la Red de Investigación Cardiovascular del Instituto de Salud Carlos III (RD12/0042/0069, RD12/0042/0049, RD12/0042/0029, RD12/0042/0069, RD12/0042/0021, RD12/0042/0021 RD12/0042/0066) y el Fondo Europeo de Desarrollo Regional (Una manera de hacer Europa).
CONFLICTO DE INTERESESNinguno.
| María Teresa Tomé-Esteban; St. George's, University of London, Londres, Reino Unido |
| Andrea Mazzanti; Fondazione Salvatore Maugeri, Pavia, Italia |
| Luis Rocha-Lopes; Hospital García de Orta, Almada, Portugal |
| Adrián Fernández; Hospital Universitario Fundación Favaloro, Buenos Aires, Argentina |
| Ricardo Stein; Universidade Federal do Rio Grande do Sul, Cardiología, Porto Alegre, Brasil |
| Ivonne J. Cárdenas-Reyes; Universidad del Rosario, Bogotá, Colombia |
| Alicia Beatriz Aguilera-Tapia; Instituto Nacional de Toxicología y Ciencias Forenses, Madrid, España |
| María Paz Suárez-Mier; Instituto Nacional de Toxicología y Ciencias Forenses, Madrid, España |
| Lorenzo Monserrat-Iglesias; Instituto de Investigación Biomédica de A Coruña, A Coruña, España |