La taquicardia ventricular polimórfica catecolaminérgica es una enfermedad maligna que se debe a mutaciones en las proteínas que controlan la homeostasis del Ca2+. Aunque el fenotipo se caracteriza por arritmias ventriculares polimórficas desencadenadas por el estrés, no se han caracterizado plenamente las arritmias supraventriculares que en ocasiones las acompañan.
MétodosVeinticinco miembros de una familia española en la que había habido varias muertes súbitas fueron evaluados mediante electrocardiograma, pruebas de esfuerzo y prueba de desenmascaramiento con adrenalina opcionalmente. Se realizó secuenciación selectiva de RyR2 en un miembro afectado y un cribado en cascada al resto de la familia. Se generó la mutación RyR2R420Q en células HEK-293 mediante mutagénesis dirigida, con objeto de realizar estudios funcionales in vitro.
ResultadosLas pruebas de esfuerzo desenmascararon taquicardia ventricular polimórfica catecolaminérgica en 8 familiares (sensibilidad del 89%; valor predictivo positivo del 100%; valor predictivo negativo del 93%), todos ellos portadores de una mutación heterocigota RyR2R420Q, que estaba presente también en el caso probando y en una chica joven sin prueba de esfuerzo, lo que corresponde a una penetrancia del 91% al final del seguimiento. Es de destacar que en los pacientes se identificó bradicardia sinusal, arritmias auriculares y de la unión y/u ondas U gigantes tras esfuerzo. Tras la permeabilización y en las células intactas, las células que expresaban RyR2R420Q mostraron un pico de liberación de Ca2+ menor que el de las células RyR2 no mutado o wild-type. Sin embargo, a una concentración de Ca2+ intracelular fisiológica, equivalente a la concentración citosólica diastólica, las células RyR2R420Q liberaban más Ca2+ y oscilaban con mayor rapidez que las células con RyR2 no mutado o wild-type.
ConclusionesLa mutación missense RyR2R420Q se identificó en el extremo aminoterminal del gen RyR2 en esta familia muy sintomática. Es de destacar que esta mutación se asocia a bradicardia sinusal, arritmias auriculares y de la unión y ondas U gigantes. En conjunto, los estudios de expresión heteróloga funcional indican que la mutación RyR2R420Q causa un comportamiento aberrante del canal, con pérdida o ganancia de función, según cuál sea la concentración de Ca2+ intracelular citosólica.
Palabras clave
La taquicardia ventricular polimórfica catecolaminérgica (TVPC) causa un 12-56% de las muertes súbitas cardiacas o paradas cardiorrespiratorias con corazón estructuralmente normal1,2. La TVPC se manifiesta en forma de síncope o muerte súbita cardiaca desencadenados por estados adrenérgicos, con una penetrancia estimada del 80%3. El electrocardiograma en reposo es generalmente normal y los pacientes presentan arritmias ventriculares (AV) durante las pruebas de esfuerzo (PE) o la infusión de catecolaminas1,4. La patogenia y la prevalencia real de las arritmias no ventriculares (como la disfunción sinusal, el marcapasos auricular errante, las arritmias de la unión y la fibrilación y flutter auriculares) que se ha descrito ocasionalmente en los pacientes con TVPC continúan sin estar claras3,5–9.
Las mutaciones suelen afectar al gen que codifica el receptor de rianodina cardiaco (RyR2), pero otros genes se han involucrado también en la patogenia de esta enfermedad10–14. El control anormal de la concentración de Ca2+ intracelular ([Ca2+]i) promueve un aumento de la liberación diastólica de Ca2+, que genera a su vez pospotenciales tardíos y con ello actividad desencadenada que es la base fisiopatológica de las AV en esta enfermedad3,15. Sin embargo, los mecanismos precisos involucrados podrían diferir según la mutación específica existente en cada caso en la proteína RyR216–18. Se presenta aquí la caracterización exhaustiva de una familia amplia con TVPC, haciendo hincapié en las manifestaciones electrocardiográficas de la enfermedad. Además, se presenta el estudio funcional in vitro que analiza el mecanismo de la disfunción del canal RyR2R420Q.
MÉTODOSEn una familia con 4 casos de muerte súbita, se llevó a cabo una evaluación familiar con un protocolo acorde con lo establecido en la Declaración de Helsinki y aprobado previamente por el comité de ética de investigación local. Se obtuvo el consentimiento informado de todos los individuos.
Estudio diagnóstico clínicoEl estudio diagnóstico en el individuo III:10 incluyó electrocardiograma, ecocardiografía, obtención de muestras de sangre y PE con ejercicio máximo (protocolo de Bruce). Una vez diagnosticado el fenotipo de TVPC durante la PE en este individuo, se ofreció un cribado en cascada a los demás miembros de la familia, que incluyó un test de adrenalina en los adultos con una PE no diagnóstica de TVPC que eran hijos de un individuo afectado. Se registró el cociente entre la amplitud máxima de onda U respecto a la onda T en esfuerzo. La bradicardia sinusal se definió como una frecuencia cardiaca < 60 lpm por encima de los 14 años de edad o inferior al percentil 2 ajustado respecto a la edad para los niños más pequeños7. La TVPC se diagnosticó en los casos en los que se producía una muerte súbita cardiaca, AV polimórfica o extrasístoles ventriculares frecuentes (> 10/min) durante una PE o un test de adrenalina (fenotipo+)19.
Estudio diagnóstico genéticoSe obtuvo ADN de muestras de sangre total (familiares) o de muestras de miocardio incluidas en parafina (probando). El análisis genético selectivo de RyR2 (secuenciación directa de los exones 3, 8, 14, 15, 37, 44-50, 83, 87-105, y regiones intrónicas adyacentes, acceso a GenBank número NM_001035) con un secuenciador ABI Prism 3100 (Applied Biosystems) en el individuo III:10 identificó la mutación RyR2R420Q en el exón 14, y en los demás familiares se secuenció únicamente dicho fragmento (se consideró genotipo+ a los portadores de la mutación). Dado que el síndrome de Andersen Tawil, debido a mutaciones del gen KCNJ2, se caracteriza por un QTc normal o casi normal, ondas U gigantes y AV relacionadas con ejercicio polimórficas, se secuenció también este gen en los individuos con TVPC.
Generación de constructos de RyR2 no mutado o wild-type y RyR2R420QSe realizó una mutagénesis dirigida de RyR2 para crear el constructo RyR2R420Q20. De forma resumida, se utilizó un plásmido que codificaba el extremo aminoterminal de la RyR cardiaca humana (Genbank X98330) para la mutagénesis dirigida (Stratagene, ChickChange kit) empleando el oligonucleótido 5’-CAGCCCGAGTTATCCAGAGCACAGTCTTCC-3’. El constructo final pRyRR420Q se generó con la digestión de SanDI/SpeI y se introdujo en un pcDNA3 (Invitrogen) que contenía la proteína RyR2 completa marcada con fluorescencia verde.
Análisis de Western blotSe realizó una transfección en células HEK-293 (European Collection of Cell Culture Agency, Salisbury, Reino Unido)20–22. Las fracciones proteicas (100 μg) se resuspendieron en tampón de carga de SDS-PAGE, y las proteínas se separaron en un gel de SDS-PAGE al 4% reforzado con agarosa al 0,5% (para la RyR)21. Las proteínas procedentes de los geles al 4% se transfirieron electroforéticamente a una membrana de polivinilidendifluoruro (Immobilon-P, Millipore) utilizando un sistema de transferencia semiseco (Trans-Blot SD, Bio-Rad) a 22 V durante 4 h. Se aplicó un anticuerpo primario específico para la isoforma de RyR2 (Ab1093, anticuerpo policlonal de conejo generado contra los residuos 4454-4474 de la RyR2) a una dilución de 1:1.000 durante una noche a 4°C. Se visualizaron las bandas de proteínas inmunorreactivas mediante una detección de quimioluminiscencia intensificada (ECL, Amersham Biosciences).
Técnica de imagen del Ca2+ intracelularLas células HEK-293 se sembraron en placas de Petri de fondo de vidrio recubiertas de polilisina D (MatTek; Ashland, Estados Unidos) y se realizó una transfección con plásmidos de RyR2 no mutado o wild-type (RyR2WT) y RyR2R420Q marcados con fluorescencia20,22. Se permeabilizaron las células para el control de la concentración de [Ca2+]i con saponina (0,01%)12. La solución interna contenía (mmol/l): K-aspartato, 120; HEPES, 10; MgATP, 2; EGTA, 0,5; Na fosfocreatina, 10; creatincinasa, 5 U/ml; MgCl2, 0,75;dextrano, 8%; saponina (0,01%), pH 7,2. Tras la permeabilización, se perfundieron las células con la misma solución interna sin saponina, pero con 15 μM de sal fluo-4-pentapotasio y la [Ca2+] conocida (10–1 – 105 nM) que se obtuvo emplendo diferentes cocientes de CaCl2:EGTA calculados con el Maxchelator23. La concentración de EGTA se mantuvo constante en 0,5mM. Dado que las células HEK-293 no son excitables, se activó la RyR2 con cafeína 5 mM y se registraron las [Ca2+]i resultantes por microscopio confocal (Zeiss LSM 510, objetivo de inmersión en agua ×63, apertura numérica, 1,2) a 394 ms/fotograma. Se realizaron también experimentos en células HEK-293 intactas según lo descrito para otros constructos de RyR224,25. En este caso, las células se cargaron con fluo-4 AM (éster acetoximetilo). Luego se perfundieron las células con una solución externa que contenía (Mm): NaCl, 150; KCl, 5,4; CaCl2, 1,5; MgCl2, 1; glucosa, 10; ácido 4-(2-hidroxietil)-1-piperacinaetanosulfónico, 5 (pH 7,4). Se registraron las oscilaciones espontáneas de la [Ca2+]i empleando microscopio confocal. Tanto en las células permeabilizadas como en las intactas, se provocó la excitación de fluorescencia de fluo-4 con un láser de Ar (488 nm) y se registró la emisión de fluorescencia > 505 nm. El análisis de las imágenes se realizó con el programa informático Zeiss LSM 510 v2.8. Se aplicó una sustracción de la fluorescencia de fondo de proteína fluorescente verde intensificada. Los valores de F (fluorescencia) se normalizaron para la F0 (fluorescencia basal, determinada antes de aplicar la cafeína o entre dos oscilaciones) con objeto de obtener el cociente de fluorescencia (F/F0).
Análisis estadísticoLas variables clínicas continuas se expresan en forma de media ± desviación estándar; las variables de laboratorio continuas, en forma de media ± error estándar de la media, y se utilizaron pruebas de la t de Student para la comparación de los grupos. Las variables dicotómicas se expresan en forma de porcentajes y se aplicó la prueba de la χ2 para la comparación de los grupos (con corrección exacta de Fisher cuando era aplicable). Un valor de p bilateral < 0,05 se consideró estadísticamente significativo. En los análisis, se utilizó el programa informático SPSS 12.0 (SPSS, Inc.; Chicago, Illinois, Estados Unidos).
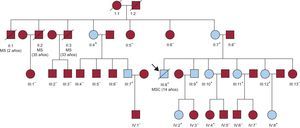
RESULTADOSAntecedentes familiares de muerte súbita cardiacaEl probando (figura 1, III:8), un varón de 14 años con antecedentes de síncopes de esfuerzo inexplicados, sufrió una muerte súbita relacionada con la práctica deportiva, y el examen post mórtem no identificó alteraciones. Otros tres familiares habían fallecido de forma súbita siendo jóvenes (uno de ellos, el individuo II:2, durante el ejercicio y con un síncope previo inexplicado), pero no se realizó autopsia a ninguno de ellos.
 al final del seguimiento. La flecha señala el caso probando inicial. Los signos más y menos indican individuos positivos y negativos para la mutación respectivamente. MS: muerte súbita, sin autopsia; MSC: muerte súbita cardiaca, sin enfermedad estructural en la autopsia. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Árbol genealógico familiar. Cuadrados: varones. Círculos: mujeres. Símbolos tachados: individuos fallecidos. Símbolos azules; taquicardia ventricular polimórfica catecolaminérgica fenotipo+ (con muerte súbita cardiaca o arritmias ventriculares relacionadas con el esfuerzo) al final del seguimiento. La flecha señala el caso probando inicial. Los signos más y menos indican individuos positivos y negativos para la mutación respectivamente. MS: muerte súbita, sin autopsia; MSC: muerte súbita cardiaca, sin enfermedad estructural en la autopsia. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
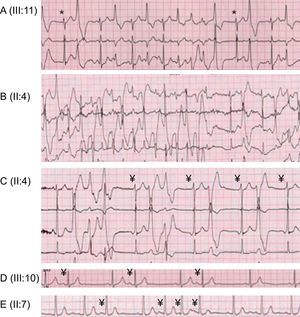
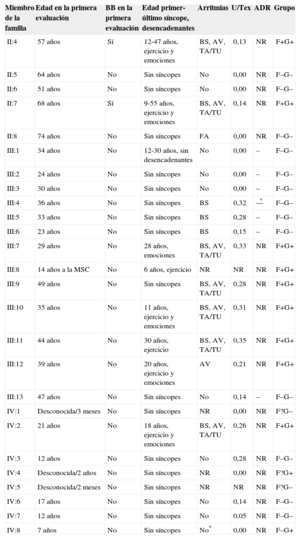
Quince años después del evento inicial, se llevó a cabo la evaluación de 25 familiares vivos del probando (tabla). Se descartó la presencia de una cardiopatía estructural con una ecocardiografía ordinaria y, en el caso del paciente III:10, también con una resonancia magnética cardiaca. No se pudo evaluar con una PE ordinaria a 3 de los familiares debido a su corta edad (fenotipo desconocido). Solo uno (IV:4) de ellos presentó genotipo+, y la monitorización electrocardiográfica no mostró ninguna AV (ni en la evaluación inicial sin fármacos a los 2 años de edad ni con bloqueadores beta en dosis ajustadas al peso a los 4 años de edad). En los 22 familiares restantes, la PE permitió establecer el diagnóstico clínico de TVPC en 8 de ellos (fenotipo+). En la PE se observó, de manera característica, que tras un aumento variable de la frecuencia cardiaca aparecían progresivamente extrasístoles ventriculares a un umbral medio de frecuencia cardiaca de 103 ± 24 (62-130) lpm, primero aisladas y monomórficas y luego con bigeminismo, polimórficas, en dobletes y también en salvas de taquicardia ventricular polimórfica no sostenida en el 50% de los individuos con fenotipo+ a un umbral medio de frecuencia cardiaca de 121 ± 10 (109-131) lpm. Finalmente, las extrasístoles ventriculares desaparecían progresivamente durante el periodo de recuperación. Tan solo un individuo presentó una taquicardia ventricular bidireccional. Es de destacar que estas arritmias no fueron en ningún caso sostenidas ni se asociaron a síncopes (figura 2). De los 13 individuos en riesgo, el test de adrenalina fue negativo en 7, lo cual concuerda con los anteriores resultados negativos de sus PE, y se desestimó en 4 niños (IV:3 y IV:6-8) y 2 adultos (II:5 y II:6), a la espera de los resultados genéticos.
Estudio diagnóstico familiar inicial
| Miembro de la familia | Edad en la primera evaluación | BB en la primera evaluación | Edad primer-último síncope, desencadenantes | Arritmias | U/Tex | ADR | Grupo |
|---|---|---|---|---|---|---|---|
| II:4 | 57 años | Sí | 12-47 años, ejercicio y emociones | BS, AV, TA/TU | 0,13 | NR | F+G+ |
| II:5 | 64 años | No | Sin síncopes | No | 0,00 | NR | F–G– |
| II:6 | 51 años | No | Sin síncopes | No | 0,00 | NR | F–G– |
| II:7 | 68 años | Sí | 9-55 años, ejercicio y emociones | BS, AV, TA/TU | 0,14 | NR | F+G+ |
| II:8 | 74 años | No | Sin síncopes | FA | 0,00 | NR | F–G– |
| III:1 | 34 años | No | 12-30 años, sin desencadenantes | No | 0,00 | – | F–G– |
| III:2 | 24 años | No | Sin síncopes | No | 0,00 | – | F–G– |
| III:3 | 30 años | No | Sin síncopes | No | 0,00 | – | F–G– |
| III:4 | 36 años | No | Sin síncopes | BS | 0,32 | –* | F–G– |
| III:5 | 33 años | No | Sin síncopes | BS | 0,28 | – | F–G– |
| III:6 | 23 años | No | Sin síncopes | BS | 0,15 | – | F–G– |
| III:7 | 29 años | No | 28 años, emociones | BS, AV, TA/TU | 0,33 | NR | F+G+ |
| III:8 | 14 años a la MSC | No | 6 años, ejercicio | NR | NR | NR | F+G+ |
| III:9 | 49 años | No | Sin síncopes | BS, AV, TA/TU | 0,28 | NR | F+G+ |
| III:10 | 35 años | No | 11 años, ejercicio y emociones | BS, AV, TA/TU | 0,31 | NR | F+G+ |
| III:11 | 44 años | No | 30 años, ejercicio | BS, AV, TA/TU | 0,35 | NR | F+G+ |
| III:12 | 39 años | No | 20 años, ejercicio y emociones | AV | 0,21 | NR | F+G+ |
| III:13 | 47 años | No | Sin síncopes | No | 0,14 | – | F–G– |
| IV:1 | Desconocida/3 meses | No | Sin síncopes | NR | 0,00 | NR | F?G– |
| IV:2 | 21 años | No | 18 años, ejercicio y emociones | BS, AV, TA/TU | 0,26 | NR | F+G+ |
| IV:3 | 12 años | No | Sin síncopes | No | 0,28 | NR | F–G– |
| IV:4 | Desconocida/2 años | No | Sin síncopes | NR | 0,00 | NR | F?G+ |
| IV:5 | Desconocida/2 meses | No | Sin síncopes | NR | NR | NR | F?G– |
| IV:6 | 17 años | No | Sin síncopes | No | 0,14 | NR | F–G– |
| IV:7 | 12 años | No | Sin síncopes | No | 0,05 | NR | F–G– |
| IV:8 | 7 años | No | Sin síncopes | No* | 0,00 | NR | F–G+ |
+: positivo; −: negativo; ?: desconocido; ADR: provocación de adrenalina; AV: arritmias ventriculares; BB: bloqueadores beta; BS: bradicardia sinusal; F: fenotipo taquicardia ventricular polimórfica catecolaminérgica; G: genotipo RyR2R420Q; MSC: muerte súbita cardiaca; NR: no realizado; PE: pruebas de esfuerzo; TA/TU: taquiarritmias auriculares y/o de la unión; U/Tex: cociente entre la amplitud máxima de onda U respecto a la onda T en esfuerzo.
. B: taquicardia ventricular no sostenida bidireccional. C: extrasístoles ventriculares, taquicardia ventricular polimórfica no sostenida, doblete ventricular polimórfico, bigeminismo ventricular monomórfico y ritmo auricular ectópico (¥). D: bigeminismo auricular (¥). E: extrasístoles auriculares aisladas y en salvas (¥).")
Arritmias ventriculares y no ventriculares durante las pruebas de esfuerzo. A: bigeminismo ventricular monomórfico y salvas de taquicardia ventricular polimórfica no sostenida (*escape de la unión). B: taquicardia ventricular no sostenida bidireccional. C: extrasístoles ventriculares, taquicardia ventricular polimórfica no sostenida, doblete ventricular polimórfico, bigeminismo ventricular monomórfico y ritmo auricular ectópico (¥). D: bigeminismo auricular (¥). E: extrasístoles auriculares aisladas y en salvas (¥).
Se identificó una mutación missense heterocigota en RyR2 (1259 G>A, R420Q) en el individuo III:10. El cribado en cascada para esa mutación identificó a un total de 11 individuos con genotipo+: los 8 con fenotipo+, una joven con fenotipo– (IV:8), una joven de fenotipo desconocido (IV:4) y también el probando inicial (III:8). Todos los familiares fueron clasificados según su fenotipo y su genotipo (+, – o desconocido) (tabla). No se identificaron mutaciones de KCNJ2.
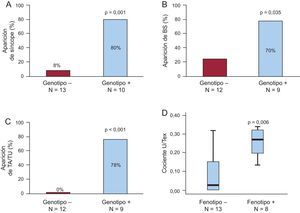
Considerando como pacientes a los portadores de mutación, la PE en la primera evaluación mostró una sensibilidad del 89%, un valor predictivo positivo del 100% y un valor predictivo negativo del 93%. La única PE con un resultado falso negativo se produjo en una niña de 7 años de edad con genotipo+ (IV:8) asintomática que presentó una conversión a fenotipo+ pese a usar bloqueadores beta 2 años después. Por consiguiente, el rendimiento general de la PE aumentó a hasta un 100%. La penetrancia de la enfermedad aumentó del 82 al 91% al final del seguimiento, al considerar los resultados clínicos (síncope y/o muerte súbita cardiaca) y de la PE en los pacientes con genotipo+. Es de destacar que la aparición de síncopes se asoció al estado de portador de mutación en los individuos evaluables > 6 años de edad (8 síncopes desencadenados con estrés/10 pacientes con genotipo+ frente a 1 síncope sin desencadenante estresante previo/13 individuos de genotipo–; p = 0,001) (tabla, figura 3A).

Manifestaciones clínicas. A: aparición de síncope; no se tuvo en cuenta a los niños de edad inferior a la del primer síncope de la familia, es decir, 6 años. B: aparición de bradicardia sinusal. C: arritmias auriculares y de la unión. D: comparación del cociente entre la amplitud máxima de onda U respecto a la onda T en esfuerzo según el fenotipo; para B-D, solamente se consideró a los individuos en ritmo sinusal y con pruebas de esfuerzo disponibles. +: positivo; –: negativo; BS: bradicardia sinusal; TA/TU: taquiarritmias auriculares y/o de la unión; U/Tex: cociente entre la amplitud máxima de onda U respecto a la onda T en esfuerzo.
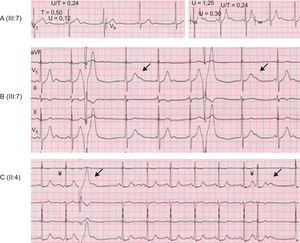
Se identificaron arritmias sinusales, auriculares, de la unión y AV en los pacientes con TVPC tanto en los electrocardiogramas en reposo como en los obtenidos durante el ejercicio. De los individuos evaluables (edad > 6 años, en ritmo sinusal y con una PE disponible), se recogieron las arritmias no ventriculares (n = 21; 9 genotipo+, 12 genotipo negativo). Se observó que la incidencia de bradicardia sinusal era superior (tabla, figura 3B) en los pacientes con genotipo+ que entre los de genotipo– (el 78 frente al 25%; odds ratio = 10,5; p = 0,030). Las taquiarritmias auriculares y/o de la unión incluyeron extrasístoles auriculares, bigeminismo auricular, taquicardia auricular no sostenida, ritmo acelerado auricular y de la unión y ritmo de escape de la unión. Se detectaron taquiarritmias auriculares y/o de la unión en 7 de 9 pacientes con genotipo+ y en ninguno de genotipo– (tabla, figura 3C). El cociente entre la amplitud máxima de onda U respecto a la onda T en esfuerzo fue significativamente superior en los individuos con fenotipo+ que entre los de fenotipo– (figura 3D). Aunque el solapamiento de los datos impidió la identificación de un punto de corte exacto. Es de destacar que se observaron ondas U gigantes tras ejercicio en varios pacientes con TVPC (figura 4). No se detectaron ondas U alternantes ni cambios de polaridad de la onda U postextrasistólica.
 y tras ejercicio (derecha); un intervalo PR corto y fijo indica una conducción auriculoventricular acelerada (izquierda). B: aumento transitorio de la amplitud de la onda U en el primer latido tras una extrasístole ventricular (flechas) en ejercicio; un ritmo de la unión compite con un ritmo sinusal. C: aumento transitorio de la amplitud de la onda U con el ejercicio, asociado a extrasístoles auriculares (flecha de la derecha) o ventriculares (flecha de la izquierda).")
Características de la onda U en los pacientes con taquicardia ventricular polimórfica catecolaminérgica. A: onda U en reposo (izquierda) y tras ejercicio (derecha); un intervalo PR corto y fijo indica una conducción auriculoventricular acelerada (izquierda). B: aumento transitorio de la amplitud de la onda U en el primer latido tras una extrasístole ventricular (flechas) en ejercicio; un ritmo de la unión compite con un ritmo sinusal. C: aumento transitorio de la amplitud de la onda U con el ejercicio, asociado a extrasístoles auriculares (flecha de la derecha) o ventriculares (flecha de la izquierda).
Se alcanzó la dosis máxima tolerada de bloqueadores beta en los individuos con genotipo+. Se implantaron cinco desfibriladores automáticos a pacientes con fenotipo+ que presentaban AV frecuentes a pesar del tratamiento con bloqueadores beta máximo (uno de ellos, el individuo IV:2, presentaba también presíncopes desencadenados por estrés). En ese momento, no había todavía publicaciones respecto al papel de la flecainida en la TVPC4. Desde ese momento no se ha registrado en la evolución ninguna descarga de los desfibriladores automáticos (media de seguimiento, 23,3 meses).
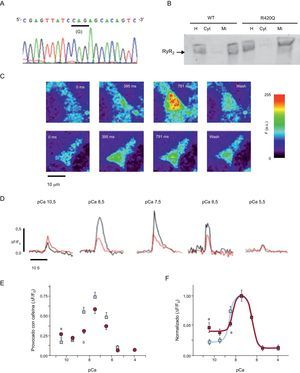
Modelo celular de RyR2R420Qin vitroCon objeto de verificar si había una alteración de la función del canal RyR2R420Q, se generó la mutación y se expresó en células HEK-293 (figura 5A). Las proteínas inmunorreactivas específicas de RyR2 expresadas se localizaron en la membrana del retículo sarcoplásmico (figura 5B). En la figura 5C se muestran ejemplos de imágenes confocales de células HEK-293 permeabilizadas con expresión de RyR2WT y RyR2R420Q tras la adición de cafeína a [Ca2+]i 10−7,5 M. Además, las imágenes registradas de los perfiles de fluorescencia a diversas [Ca2+]i se representan gráficamente en la figura 5D. La liberación de Ca2+ tanto con RyR2WT como con RyR2R420Q mostró una curva en forma de campana al representarla gráficamente en función de la [Ca2+]i citosólica, con una reducción del 34% en el pico de liberación de Ca2+ en las células con expresión de RyR2R420Q (figura 5E) a una pCa 7,5. Se descartó que un grado diferente de expresión de RyR2 en cada grupo celular para explicar esta observación al determinar una fluorescencia de la proteína RyR2 marcada con fluorescencia verde similar en las células RyR2WT y RyR2R420Q (F, en unidades arbitrarias, 50,4 ± 1,6 en 195 células RyR2WT frente a 50,1 ± 1,5 en 241 células con expresión de RyR2R420Q; sin diferencias significativas). Sin embargo, al valor más bajo de pCa evaluado, se observó un aumento significativo de la liberación de Ca2+ provocada por cafeína en las células RyR2R420Q (pCa, 10,5) (figuras 5D y E). Para calcular la EC50(half maximal effective concentration) para la curva de activación de Ca2+ y la IC50(half maximal inhibitory concentration) para la curva de inactivación del Ca2+, se normalizó la fluorescencia causada por cafeína respecto a su máximo, que resultó suceder a la misma [Ca2+]i (pCa, 7,5) en ambos grupos (figura 5F). La EC50 fue inferior en las RyR2R420Q que en las RyR2WT (2,66 ± 0,13 en las RyR2R420Q frente a 4,98±5,15nM en las RyR2WT; sin diferencias significativas). A una [Ca2+]i superior, RyR2 se inactivó con un patrón similar, según lo observado en la parte descendente de las curvas ajustadas (IC50, 365,5 ± 50,5 en RyR2WT frente a 339,5 ± 58,4 nM en RyR2R420Q; sin diferencias significativas).
![Evaluación funcional in vitro de la mutación RyR2R420Q. A: electroferograma que confirma la introducción del punto de mutación G1380A que da lugar a la conversión de arginina (A) a glutamina (G), RyR2R420Q, en el constructo del plásmido. B: análisis Western blot de RyR2 procedente de células HEK-293 transfectadas que confirma que las proteínas RyR2WT y RyR2R420Q se expresan en el homogeneizado y en las fracciones microsomales, pero no en el citosol. La flecha señala la banda de 595 kDa (RyR2 marcada con proteína fluorescente verde intensificada). C: imágenes confocales de los cambios del calcio en células HEK-293 permeabilizadas que expresan RyR2WT (arriba) y RyR2R420Q (abajo) y sumergidas en una solución interna con un contenido de [Ca2+]i = 10-7.5 M y cafeína 5 mM. D: ejemplos representativos de perfiles de fluorescencia obtenidos de células HEK-293 permeabilizadas con expresión de RyR2WT (negro) y RyR2R420Q (rojo) y sumergidas en soluciones que contienen diferentes [Ca2+]i y estimuladas con cafeína 5 mM. E: promedio de liberación de calcio inducida por cafeína (valores máximos normalizados para el cociente de fluorescencia basal) en las células HEK-293 con expresión de RyR2WT (cuadrados) y RyR2R420Q (círculos) a diversas [Ca2+]i; los datos se expresan en forma de media ± error estándar de la media (n = 20-50 células por grupo). F: promedio de fluorescencia deproteína fluorescente verde intensificada-RyR2 observada en las células HEK-293 con expresión de RyR2WT (n = 195) y RyR2R420Q (n = 241). △: incremento; Cyt: citosol; eGFP: proteína fluorescente verde intensificada; F: fluorescencia; F0: fluorescencia basal; H: homogeneizado; Mi: fracciones microsomales. ap < 0,05, RyR2R420Q frente a RyR2WT. bp < 0,001. Esta figura se muestra a todo color solo en la versión electrónica del artículo.](https://static.elsevier.es/multimedia/03008932/0000006800000005/v1_201504231253/S0300893214004278/v1_201504231253/es/main.assets/gr5.jpeg?xkr=eyJpdiI6IklXRU5kdVV1MkZ3VGIzVGVSM2dGM0E9PSIsInZhbHVlIjoiczNHZHAwUVRFR28rRFoyYXJIb3hEc3RXbkRhUlN0SWNQSG1VMk1zNmJiM0lCOGZPRGUzcUplN1VwSloxbVovWlZBb3hEc0R4cUtwUmFTbytJa2NpSFVtSjAyVmJZMzJLNndmVGtiWm82SUhPSTZMY2h0RjdrK0p3aExzZ0JEZUdzaUlmMTJCM2hXRmxNOTUwcndHTU85cHpQV2pJTEFtOTBSMGJQQ09HVkxLTVAyZ25pNVFQWEQrT01rWjRNUGcxRHV4a25OR0ZRaUlyc1Q0SnF2cE5qMWtpMlFpcEV6U2h2T1g1Ym4rcnYxNVd1RHp3TWZXMzA1cnlPNVpZVlg2bUpudnZEaWhOdzBJd0hZMjZqUzVVOU4rK2h5WGxXM3hxYWgxSFZrcnYyV2s9IiwibWFjIjoiMTVmY2NhOTY2ODhkMDliNWVhMjBiZDM4MGZhMDdkZWFkNGEwMDRhMzkyNjQ0YTgxZmM1ZGIzZDg2Yzg1MTYxNyIsInRhZyI6IiJ9 "Evaluación funcional in vitro de la mutación RyR2R420Q. A: electroferograma que confirma la introducción del punto de mutación G1380A que da lugar a la conversión de arginina (A) a glutamina (G), RyR2R420Q, en el constructo del plásmido. B: análisis Western blot de RyR2 procedente de células HEK-293 transfectadas que confirma que las proteínas RyR2WT y RyR2R420Q se expresan en el homogeneizado y en las fracciones microsomales, pero no en el citosol. La flecha señala la banda de 595 kDa (RyR2 marcada con proteína fluorescente verde intensificada). C: imágenes confocales de los cambios del calcio en células HEK-293 permeabilizadas que expresan RyR2WT (arriba) y RyR2R420Q (abajo) y sumergidas en una solución interna con un contenido de [Ca2+]i = 10-7.5 M y cafeína 5 mM. D: ejemplos representativos de perfiles de fluorescencia obtenidos de células HEK-293 permeabilizadas con expresión de RyR2WT (negro) y RyR2R420Q (rojo) y sumergidas en soluciones que contienen diferentes [Ca2+]i y estimuladas con cafeína 5 mM. E: promedio de liberación de calcio inducida por cafeína (valores máximos normalizados para el cociente de fluorescencia basal) en las células HEK-293 con expresión de RyR2WT (cuadrados) y RyR2R420Q (círculos) a diversas [Ca2+]i; los datos se expresan en forma de media ± error estándar de la media (n = 20-50 células por grupo). F: promedio de fluorescencia deproteína fluorescente verde intensificada-RyR2 observada en las células HEK-293 con expresión de RyR2WT (n = 195) y RyR2R420Q (n = 241). △: incremento; Cyt: citosol; eGFP: proteína fluorescente verde intensificada; F: fluorescencia; F0: fluorescencia basal; H: homogeneizado; Mi: fracciones microsomales. ap < 0,05, RyR2R420Q frente a RyR2WT. bp < 0,001. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Evaluación funcional in vitro de la mutación RyR2R420Q. A: electroferograma que confirma la introducción del punto de mutación G1380A que da lugar a la conversión de arginina (A) a glutamina (G), RyR2R420Q, en el constructo del plásmido. B: análisis Western blot de RyR2 procedente de células HEK-293 transfectadas que confirma que las proteínas RyR2WT y RyR2R420Q se expresan en el homogeneizado y en las fracciones microsomales, pero no en el citosol. La flecha señala la banda de 595 kDa (RyR2 marcada con proteína fluorescente verde intensificada). C: imágenes confocales de los cambios del calcio en células HEK-293 permeabilizadas que expresan RyR2WT (arriba) y RyR2R420Q (abajo) y sumergidas en una solución interna con un contenido de [Ca2+]i = 10-7.5 M y cafeína 5 mM. D: ejemplos representativos de perfiles de fluorescencia obtenidos de células HEK-293 permeabilizadas con expresión de RyR2WT (negro) y RyR2R420Q (rojo) y sumergidas en soluciones que contienen diferentes [Ca2+]i y estimuladas con cafeína 5 mM. E: promedio de liberación de calcio inducida por cafeína (valores máximos normalizados para el cociente de fluorescencia basal) en las células HEK-293 con expresión de RyR2WT (cuadrados) y RyR2R420Q (círculos) a diversas [Ca2+]i; los datos se expresan en forma de media ± error estándar de la media (n = 20-50 células por grupo). F: promedio de fluorescencia deproteína fluorescente verde intensificada-RyR2 observada en las células HEK-293 con expresión de RyR2WT (n = 195) y RyR2R420Q (n = 241). △: incremento; Cyt: citosol; eGFP: proteína fluorescente verde intensificada; F: fluorescencia; F0: fluorescencia basal; H: homogeneizado; Mi: fracciones microsomales. ap < 0,05, RyR2R420Q frente a RyR2WT. bp < 0,001. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
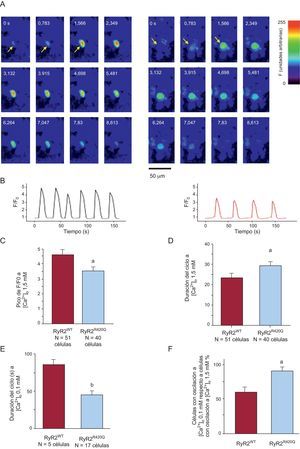
Las células HEK-293 intactas con transfección con plásmidos de RyR2WT o RyR2R420Q se utilizaron para analizar las oscilaciones espontáneas de la [Ca2+]i (figuras 6A y B). Es interesante señalar que, a [Ca2+]o fisiológicas (1,5 mM) [Ca2+]i, las oscilaciones registradas en las células RyR2R420Q fueron de menor amplitud (figura 6C), de menor duración (7,6 ± 0,5 ms en 51 células RyR2WT frente a 5,9 ± 0,5 ms en 40 células RyR2R420Q; p < 0,05), y tuvieron una duración del ciclo mayor que las oscilaciones de las células RyR2WT (figura 6D). Es llamativo que estas alteraciones no se debieran a una disminución de la carga de Ca2+ del retículo sarcoplásmico, que de hecho fue similar en los dos grupos de células (F/F0, 3,8 ± 0,3 en 15 células frente a 3,6 ± 0,3 en 13 células RyR2R420Q, sin diferencias significativas). En un grupo de células, registramos primero las oscilaciones espontáneas a 1,5 mM [Ca2+]o y luego redujimos la [Ca2+]o a 0,1 mM [Ca2+]o. Esta maniobra produjo una supresión de la actividad automática en un porcentaje de las células HEK-293 superior en las RyR2WT que en las RyR2R420Q, reflejado en una frecuencia superior (o menor duración del ciclo) de las oscilaciones espontáneas (figura 6E) y un porcentaje más alto de células con expresión de RyR2R420Q que mostraban oscilación (figura 6F), todo ello en consonancia con los resultados de este estudio en las células permeabilizadas a los valores más bajos de [Ca2+]i analizados.
![Las oscilaciones espontáneas del Ca2+ en las células HEK-293 que expresan RyR2R420Q son menores y más lentas, pero se convierten en más rápidas que las de RyR2WT a un valor inferior de [Ca2+]o. A: serie temporal de imágenes confocales en células HEK-293 intactas con expresión de RyR2WT (izquierda) y RyR2R420Q (derecha). B: perfiles de fluorescencia representativos en células HEK-293 intactas que expresan RyR2WT (izquierda) y RyR2R420Q (derecha), a una [Ca2+]o de 1,5 mM. C: amplitud de las oscilaciones de Ca2+ a una [Ca2+]o de 1,5 mM. D: igual que en C, pero para la duración del ciclo entre oscilaciones consecutivas del Ca2+. E: igual que en D, pero a una [Ca2+]o de 0,1 mM. F: porcentaje de células que oscilan a una [Ca2+]o 0,1 mM respecto a las células que oscilan a una [Ca2+]o 1,5 mM. RyR2R420Q frente a RyR2WT. ap<0,05. bp<0,01. Esta figura se muestra a todo color solo en la versión electrónica del artículo.](https://static.elsevier.es/multimedia/03008932/0000006800000005/v1_201504231253/S0300893214004278/v1_201504231253/es/main.assets/gr6.jpeg?xkr=eyJpdiI6Im05cWliYUlSb0tBL2RhRUE5UGlnZ2c9PSIsInZhbHVlIjoiMVYzTHh6ek5jT2JlK0FkQXJEdXE1dlE2WEtYMlhpNGI3OFRhSmlMeHNzUXNQVGtzcm81ZU8rbzNvd1NMNGJBSkIyd2o1VzFtVURDaS9wWExIQytVWFdOMmc1NjdTYzNHK2JYb3lYWTBJUFl3Mk1UdlE3dzNIQmtUN2lJZVFIKzBIcTF0WTU3QW85VnJtUnVEaG9TWlREcm5aWTVqay9aeUxPN3ZaZW8vQWtRZTBKS1l3T1BIZWRvY012YVZrZVZjSHYzZzJNZXdVbFdDRS9FNmZLVFlFVzRmb2pvc3NnZEVJeDFRdVAyZmdPMDdINDgvZEtna1RmN3RpdCtMZk5YK2hjMThrVVBEL2dxR1V0SXJsclR0Q2N5VXB1MDZTZXFvMGNSeVdiRlU5U3c9IiwibWFjIjoiYjMwNjk1ZTgyMTlhM2RlZGYxOWRmODQ5NzRhYmY0NzQzYjA3MzE4MTk4MzE3ZGY0ZTk2OTI3NWRiNGFhNmJkMyIsInRhZyI6IiJ9 "Las oscilaciones espontáneas del Ca2+ en las células HEK-293 que expresan RyR2R420Q son menores y más lentas, pero se convierten en más rápidas que las de RyR2WT a un valor inferior de [Ca2+]o. A: serie temporal de imágenes confocales en células HEK-293 intactas con expresión de RyR2WT (izquierda) y RyR2R420Q (derecha). B: perfiles de fluorescencia representativos en células HEK-293 intactas que expresan RyR2WT (izquierda) y RyR2R420Q (derecha), a una [Ca2+]o de 1,5 mM. C: amplitud de las oscilaciones de Ca2+ a una [Ca2+]o de 1,5 mM. D: igual que en C, pero para la duración del ciclo entre oscilaciones consecutivas del Ca2+. E: igual que en D, pero a una [Ca2+]o de 0,1 mM. F: porcentaje de células que oscilan a una [Ca2+]o 0,1 mM respecto a las células que oscilan a una [Ca2+]o 1,5 mM. RyR2R420Q frente a RyR2WT. ap<0,05. bp<0,01. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Las oscilaciones espontáneas del Ca2+ en las células HEK-293 que expresan RyR2R420Q son menores y más lentas, pero se convierten en más rápidas que las de RyR2WT a un valor inferior de [Ca2+]o. A: serie temporal de imágenes confocales en células HEK-293 intactas con expresión de RyR2WT (izquierda) y RyR2R420Q (derecha). B: perfiles de fluorescencia representativos en células HEK-293 intactas que expresan RyR2WT (izquierda) y RyR2R420Q (derecha), a una [Ca2+]o de 1,5 mM. C: amplitud de las oscilaciones de Ca2+ a una [Ca2+]o de 1,5 mM. D: igual que en C, pero para la duración del ciclo entre oscilaciones consecutivas del Ca2+. E: igual que en D, pero a una [Ca2+]o de 0,1 mM. F: porcentaje de células que oscilan a una [Ca2+]o 0,1 mM respecto a las células que oscilan a una [Ca2+]o 1,5 mM. RyR2R420Q frente a RyR2WT. ap<0,05. bp<0,01. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Por primera vez se describe el fenotipo asociado a la mutación RyR2R420Q en una familia muy síntomatica, y destaca la elevada incidencia de arritmias no ventriculares. Los resultados in vitro confirman una disfunción del canal mutante.
El rendimiento general de la PE inicial para el diagnóstico de la TVPC (sensibilidad, 89%; valor predictivo positivo, 100%, y valor predictivo negativo, 93%) fue superior al descrito en otras series26. El comportamiento de dos pacientes jóvenes con genotipo+ (IV:4 e IV:8) indica que la penetrancia podría estar relacionada con la edad. Además, hubo bradicardia sinusal, taquiarritmias auriculares y/o de la unión y ondas U gigantes tras ejercicio en los portadores de RyR2R420Q (figuras 3 y 4). Se ha descrito ocasionalmente ondas U alternantes y cambios de polaridad de la onda U postextrasistólica en individuos con TVPC27 (como consecuencia de una alteración de la homeostasis del Ca2+), y también un aumento del cociente U/T28,29 (que se considera la contrapartida electrocardiográfica de los pospotenciales tardíos). Es de destacar que los pacientes del estudio con TVPC presentaron un aumento de la amplitud de la onda U postextrasistólica y un aumento del cociente U/T con el ejercicio, precisamente con estímulo catecolaminérgico. El origen de las arritmias supraventriculares puede atribuirse a la mutación de RyR2, ya que RyR2 se expresa en todos los miocardiocitos, incluidas las células marcapasos y los miocitos auriculares30,31. De hecho, recientemente se ha observado que la mutación RyR2R4496C fomenta un aumento del automatismo en las células ventriculares15, pero con una disminución del automatismo en las células del nódulo sinoauricular9. Así pues, los canales RyR2 mutados parecen presentar una actividad disfuncionante que depende del tipo celular.
RyR2R420Q forma parte del cluster aminoterminal e induce la sustitución del aminoácido básico de carga eléctrica alta (arginina) por un residuo aminoácido sin carga eléctrica (glutamina). Esta mutación, que se ha descrito recientemente en 4 pacientes no emparentados, por ahora carece de caracterización clínica y funcional14,32. Es de destacar que el residuo aminoacídico 420 está muy conservado en las distintas especies32 y se comporta como un punto caliente, dado que se han descrito diferentes sustituciones en el mismo punto y cerca de él (R420W3,33–35 e I419F36,37). Conviene señalar que RyR2R420Q muestra una penetrancia superior a la de RyR2R420W (el 91 frente al 25%), y no manifiesta ninguna característica propia de una miocardiopatía arritmogénica33,34.
Hasta ahora, la mayor parte de las mutaciones de la TVPC en las que se han realizado estudios funcionales se han comportado como mutaciones de ganancia de función35,38. El aumento de la sensibilidad al Ca2+ (luminal o citosólico)15,24,38,39, una alteración de la interacción entre los dominios aminoterminal y central40,41, y la disminución de la unión de FKBP12.638,39 constituyen los mecanismos más aceptados. Muy recientemente, se ha observado un efecto perturbador de la mutación RyR2R420Q en la estructura cristalina de la región aminoterminal de RyR2, puesto que produce una supresión de la fijación del cloruro, que altera su pliegue de su dominio42,43 a pesar de que, en términos lineales, el residuo aminoácido 420 esté muy lejos de la región molecular para la que se propone la intervención en la activación dependiente del Ca2+ (residuos 4485-4494)40. Se ha observado también que el extremo aminoterminal es una región estructural importante con capacidad de autotetramerizar, y puede intervenir en la regulación de la función del canal nativo44. Así pues, parece claro desde un punto de vista estructural estático que RyR2R420Q desestabiliza la interfaz entre subunidades, lo que probablemente facilite la apertura del canal42–45.
Se presenta aquí la primera evaluación inicial de la mutación RyR2R420Q desde una perspectiva funcional. En primer lugar, los resultados de este estudio reflejan claramente que la mutación RyR2R420Q se comporta como una mutación de ganancia de función tanto en las células permeabilizadas como en las intactas (figuras 5D, 6F, 6E y 6F) a una [Ca2+]i muy baja, similar a la determinada en los miocardiocitos de rata y humanos46,47. En segundo lugar, la razón de que se reduzca el pico de liberación de [Ca2+] causado por la cafeína en las células con RyR2R420Q (figura 5E) no tiene una interpretación sencilla. Podría estar relacionada con un canal hipoactivo a valores de Ca2+ citosólico superiores, pero sin una desviación significativa de la EC50. Por último, en las células RyR2R420Q intactas, las oscilaciones espontáneas mostraron una disminución de la amplitud y la frecuencia (figuras 6B y 6D). Esta disminución podría explicar la bradicardia sinusal observada en los pacientes con TVPC (figura 3B), y no depende de una menor carga de Ca2+ en el retículo sarcoplásmico, ya que esta fue normal (véase «Resultados»).
Limitaciones del estudioLos sistemas de expresión celular heterólogos son útiles para caracterizar las mutaciones de RyR2, aunque pueden tener limitaciones, ya que no permiten replicar el comportamiento eléctrico existente in vivo, en el que otras proteínas y el tono del sistema autónomo pueden interaccionar con el canal disfuncional. Así pues, los experimentos realizados en ratones transgénicos o en iPS-CM (cardiomiocitos derivados de células madre pluripotentes inducidas) aportarán conclusiones más claras.
CONCLUSIONESEste trabajo aporta una caracterización clínica novedosa y detallada de la mutación RyR2R420Q. Los análisis resaltan que las arritmias no ventriculares son frecuentes y podrían reforzar la sospecha de TVPC en un contexto clínico apropiado. Además, los análisis in vitro indican que la RyR2R420Q da lugar a un canal aberrante.
FINANCIACIÓNEste trabajo contó con el apoyo de Instituto de Salud Carlos III (PI14/01477, RD12/0042/0029), Sociedad Española de Cardiología (Proyecto de Investigación Clínica en Cardiología Dr. Pedro Zarco), Biobanco La Fe (PT13/0010/0026), Prometeo 2011/027, ANR (Agence Nationale de la Recherche) (ANR-13-BSV1-0023), y Région Île-de-France (CORDIM, COD 100256). El INSERM (Institut National de la Santé et de la Recherche Médicale) U-769 forma parte del Laboratory of excellence LERMIT (Laboratory of Excellence in Research on Medication and Innovative Therapeutics), que cuenta con el apoyo de una subvención de ANR Investissements d’avenir. Patricia Neco contó con el respaldo de la Fondation pour la Recherche Médicale y Spyros Zissimopoulos, con el de la British Heart Foundation.
CONFLICTO DE INTERESESNinguno.