
La amiloidosis hereditaria por transtirretina (ATTRv) es una enfermedad sistémica con un patrón de transmisión autosómico dominante, y se han descrito más de 100 variantes patogénicas. La prevalencia de la ATTRv difiere entre distintas regiones, y la enfermedad es endémica en determinadas zonas geográficas debido a efectos fundadores. Por ejemplo, la variante p.Val50Met es endémica en Povoa de Varzim (Portugal), Västerbotten (Suecia) y Mallorca (España)1. La variante p.Glu109Lys es la tercera mutación más frecuente en España y se define por una edad de inicio temprana, una afección mixta cardiaca y neurológica y un mal pronóstico. En un estudio reciente se detectó un efecto fundador de la variante cuyo origen se cree en el sudeste de España2,3.
La ATTRv es una enfermedad óptima para aplicar medidas de cribado por su importante repercusión en la salud, el largo periodo de latencia de la enfermedad y la disponibilidad de pruebas de cribado no invasivas y tratamientos modificadores de la enfermedad eficaces1,4. A la vista de estas características, nuestro objetivo es examinar la viabilidad de aplicar un programa de cribado en el municipio de origen de la variante p.Glu109Lys.
Para ello, se diseñó un estudio prospectivo destinado a ofrecer un cribado genético para el gen de la transtirretina (TTR) a los habitantes de Villacarrillo con riesgo de ATTRv. Los criterios de inclusión fueron tener edad entre 40 y 70 años y al menos 1 de las siguientes señales de alerta clínicas de la ATTRv: diagnóstico de insuficiencia cardiaca no explicada por una miocardiopatía isquémica o una valvulopatía, hipertrofia del ventrículo izquierdo (≥12mm) en la ecocardiografía, implante de marcapasos a causa de trastornos de la conducción, signos o síntomas de neuropatía periférica definidos por la presencia de parestesias, déficits sensitivos o dolor neuropático sin que hubiera ninguna otra enfermedad neurológica, síndrome del túnel carpiano y estenosis raquídea lumbar. Se excluyó a los pacientes con un estudio genético previo de TTR o con cualquier trastorno que les impidiera tomar una decisión informada sobre la realización de las pruebas genéticas. La identificación de los candidatos se llevó a cabo mediante un examen a distancia de las historias clínicas electrónicas del centro de atención primaria (CAP) de Villacarrillo. El personal del CAP contactó por teléfono o durante las visitas ordinarias con los pacientes que cumplían los criterios de inclusión para invitarles a participar. Los pacientes que aceptaron participar firmaron un documento de consentimiento informado, recibieron asesoramiento genético y proporcionaron una muestra de saliva para las pruebas genéticas del gen TTR. El personal médico del CAP entregó los resultados genéticos a los participantes.
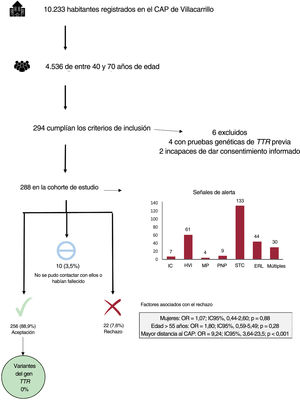
Se incluyó a todos los habitantes de Villacarrillo registrados en el CAP en febrero de 2022 (10.233 personas). Se analizaron las historias clínicas de un total de 4.536 personas de entre 40 y 70 años de edad. Tras el examen de la historia clínica, se identificó a 294 habitantes como posibles candidatos, pero se excluyó a 6 de ellos porque ya se les había hecho pruebas genéticas de TTR (n=4) o porque no eran capaces de tomar una decisión informada (n=2). Formaron la cohorte de estudio final 288 pacientes (el 6,4% de los pacientes a los que se aplicó el cribado). La media de edad fue de 59,1±7,5 años y 164 (56,9%) eran mujeres. Por lo que respecta a las señales de alerta clínicas, el síndrome del túnel carpiano fue la más frecuente (n=133, 46,2%), seguido de hipertrofia del ventrículo izquierdo (n=61, 21,2%) y estenosis raquídea lumbar (n=44, 15,3%). Un total de 30 pacientes (10,4%) presentaron 2 o más señales de alerta. Se realizaron pruebas genéticas a 256 personas (88,9%), mientras que 22 participantes (7,6%) rechazaron la participación y 10 (3,5%) no fueron incluidos porque no se pudo contactar con ellos (n=6, 2,1%) o fallecieron antes de haberlo hecho (n=4, 1,4%). En un análisis exploratorio se observó que los pacientes que no vivían en la localidad principal del municipio tenían una mayor probabilidad de rechazar el cribado (odds ratio [OR] = 9,24; IC95%, 3,64-23,5; p<0,001). Las pruebas genéticas de todos los pacientes resultaron satisfactorias, sin que se produjera ningún incidente, pero no se identificó a ningún portador de la variante p.Glu109Lys ni de ninguna otra variante patogénica (figura 1).

Diagrama de flujo de las pruebas genéticas. CAP: centro de atención primaria; ERL: estenosis raquídea lumbar; HVI: hipertrofia del ventrículo izquierdo; IC: insuficiencia cardiaca; IC95%: intervalo de confianza del 95%; MP: marcapasos; OR: odds ratio; PNP: polineuropatía; STC: síndrome del túnel carpiano; TTR: gen de la transtirretina.
Hasta donde nosotros sabemos, este es el primer estudio en el que se ha llevado a cabo un programa de cribado poblacional dirigido a una cardiopatía hereditaria dentro de una región específica de España. Nuestros resultados indican que este enfoque es viable y tiene una alta tasa de aceptación entre los posibles participantes. Consideramos que hay 2 factores que contribuyeron a producir esta alta tasa de aceptación. En primer lugar, la participación del personal médico local para contactar con los pacientes, que puede haber generado confianza. En segundo lugar, el uso de kits para muestras de saliva, en vez de métodos de extracción de sangre, facilitó la participación. Además, nuestros resultados indican también que la proximidad desempeña un papel importante en la aceptación. Nuestro trabajo muestra también que las señales de alerta de la ATTR son muy frecuentes en la población general, ya que se dieron en un 6% de los participantes de entre 40 y 70 años. Estos resultados apuntan a que los futuros proyectos diseñados para el cribado de la ATTR en la población general podrían verse beneficiados con un enfoque más dirigido y centrado en señales de alerta más específicas o en una combinación de señales de alerta frecuentes, a fin de incrementar la especificidad y alcanzar una relación coste-efectividad favorable.
Se prevé un crecimiento exponencial de los programas de cribado genético poblacional en los próximos años, gracias al acceso a pruebas genéticas de menor coste. En términos generales, los programas piloto se centran en los recién nacidos y, aunque incluyen un gran número de enfermedades, por lo general no han incluido las cardiopatías hereditarias5. Se ha propuesto un cribado genético poblacional para la ATTR, en especial en los países con gran prevalencia de población negra, ya que la variante p.Val142Ile patogénica afecta a un 3-4% de las personas negras en Estados Unidos6. Estos programas podrían basarse en la obtención automática de gran cantidad de datos (big data) a partir de las historias clínicas electrónicas, con un diseño que permita detectar señales de alerta específicas.
En resumen, a pesar del resultado negativo de nuestro proyecto de cribado, nuestra experiencia aporta una perspectiva útil acerca de la viabilidad de los programas de cribado genético y los posibles obstáculos para su aplicación en situaciones de la vida real.
FINANCIACIÓNEste trabajo fue financiado por una subvención de Alnylam Pharmaceuticals Inc. para una investigación iniciada por el investigador.
CONSIDERACIONES ÉTICASEl estudio fue aprobado por el comité de ética del Hospital Universitario Puerta de Hierro y el Hospital Universitario de Jaén y los participantes dieron su consentimiento informado. El sexo y el género no se tuvieron en cuenta según lo establecido en SAGER.
DECLARACIÓN SOBRE EL USO DE INTELIGENCIA ARTIFICIALNo se ha utilizado ninguna herramienta de inteligencia artificial en la preparación de este artículo.
CONTRIBUCIÓN DE LOS AUTORESF. de Frutos y I. Caraballo Ramos son ambos primeros autores. F. de Frutos y P. García-Pavía diseñaron el estudio. F. de Frutos, I. Caraballo Ramos, V. Martínez Chaves, A.M. Corral Azor y M.S. Berchíd Débdi obtuvieron los datos. F. de Frutos y P. García-Pavía elaboraron la versión inicial del manuscrito. I. Caraballo Ramos, V. Martínez Chaves, A.M. Corral Azor y M.S. Berchíd Débdi realizaron una revisión crítica del manuscrito. P. García-Pavía se encargó de la obtención de financiación.
CONFLICTO DE INTERESESP. García-Pavía es editor asociado de Rev Esp Cardiol; se ha seguido el procedimiento editorial establecido en la Revista para garantizar la gestión imparcial del manuscrito. El resto de los autores no tiene conflictos de intereses.