
El ST2 circulante es la isoforma soluble de un receptor de la familia de la interleucina 1 cuya primera descripción se sitúa en 1989, en relación con procesos inflamatorios de origen inmunológico1. Su estudio dentro de la patología cardiovascular se inició en 2001 cuando, la mayor expresión en cardiomiocitos sometidos a estrés mecánico, de entre 7.000 genes transcritos con función conocida, fue la de ST22.
El trabajo de Arrieta et al.3 publicado recientemente en Revista Española de Cardiología aporta nuevos hallazgos que refuerzan un cambio de visión acerca del papel del ST2 circulante en la enfermedad cardiovascular: de un biomarcador identificador de riesgo a un patógeno implicado en el desarrollo de enfermedad, en este caso en relación con la fibrosis miocárdica y el remodelado ventricular adverso en la estenosis aórtica. En el trabajo pionero de Winberg et al.2 se describió su aumento en las primeras horas tras un infarto de miocardio, y pronto fue identificado como un biomarcador de riesgo en presencia de insuficiencia cardiaca4. Durante estos más de 20 años, han sido muchas las descripciones de la asociación entre concentraciones circulantes de ST2 aumentadas y un mayor riesgo de complicaciones clínicas, en particular su asociación con una mayor mortalidad5. Durante este largo tiempo, su determinación ha pasado de inmunoanálisis manuales con amplia variabilidad a automatizarla en laboratorios clínicos con adecuados estándares de calidad, e incluso como pruebas rápidas en el punto de atención al paciente. De hecho, su medición como marcador de riesgo asociado con procesos de fibrosis se recogió en la guía estadounidense de 20136. Sin embargo, su incorporación real a la práctica clínica como marcador de riesgo ha sido muy escasa en Europa.
Nuestro grupo le prestó atención muy al inicio y aportó evidencias pioneras en relación con un peor pronóstico en pacientes con insuficiencia cardiaca, con 2 elementos relevantes: su valor complementario sobre péptidos natriuréticos y troponinas7,8 y su gran capacidad de discriminación para identificar mortalidad y muerte súbita en particular9. Por otro lado, y de modo muy relevante, diferentes trabajos han mostrado su asociación con peor remodelado miocárdico, en particular tras un infarto agudo de miocardio10. Su valor complementario sobre otros biomarcadores debe interpretarse como que no se trata solo de un simple marcador de riesgo, sino que estaría reflejando un proceso fisiopatológico diferente y, probablemente, un papel patogénico. Su potente asociación con la mortalidad y su valor predictivo en diferentes enfermedades y contextos clínicos, cuyo punto en común es la afección cardiaca y el riesgo de progresión a daño y disfunción miocárdicas, dan de nuevo relevancia a ese proceso patogénico. A fecha de hoy, la evidencia acumulada nos muestra que las concentraciones circulantes de ST2 aumentadas no solo identifican mayor riesgo en pacientes que ya sufren insuficiencia cardiaca11, sino también otras cardiopatías, el infarto en particular, o incluso afecciones extracardiacas cuando conllevan un daño miocárdico y un riesgo de afección miocárdica, como puede ser el distrés respiratorio agudo o, más recientemente, la enfermedad coronavírica de 2019 (COVID-19)5,12.
El trabajo de Arrieta et al.3 aporta hallazgos de interés. El primero añade más evidencia a la asociación entre mayor concentración circulante de ST2 y un peor remodelado miocárdico, con mayor disfunción ventricular. Este hecho ha sido bien establecido durante estos años, tanto tras infarto agudo de miocardio como en pacientes con insuficiencia cardiaca, hasta el punto de incluirse en modelos predictivos de remodelado adverso10,13. El segundo proporciona evidencia en una enfermedad valvular con riesgo de progresión al daño y disfunción miocárdicos: la estenosis aórtica. Y el último, su hallazgo más relevante, la asociación entre una mayor concentración de ST2 circulante y la magnitud de la fibrosis miocárdica evaluada por resonancia cardiaca, cuyo papel patogénico en la progresión del remodelado y el riesgo de disfunción ventricular y de arritmias ventriculares es claro y para el que actualmente se carece de tratamientos relevantes. En este sentido, llama la atención que las concentraciones elevadas (≥ 28,2 ng/ml) estuvieran indefectiblemente asociadas con presencia de fibrosis, lo que fortalece aún más esta asociación. Otro aspecto que comentar es la mayor asociación patogénica en varones, con más fibrosis y remodelado adverso. Este aspecto podría ser un artefacto por limitaciones inherentes al diseño trasversal, el pequeño tamaño muestral y el desequilibrio en variables confusoras, pero al menos respalda la necesidad de seguir avanzando en el estudio en uno y otro sexo por separado, no solo del ST2 sino de otros muchos sistemas biológicos.
Nuestro grupo, en su día pionero en mostrar la asociación entre ST2 y mayor riesgo, se ha enfocado en los últimos años en investigar los mecanismos de regulación del ST2 con la hipótesis de que, siendo un patógeno, una respuesta nociva, como otras muchas en el sistema cardiovascular, debería considerarse una diana terapéutica14. En este sentido, hemos mostrado que el ST2 soluble (sST2) circulante no tiene un origen principal en el miocardio, sino que los pulmones tienen un papel fundamental en su producción, lo cual ahonda en una interacción cardiopulmonar, así como las células endoteliales e inflamatorias15-17. Además, el estímulo a la secreción de ST2 circulante debe interpretarse como una respuesta inflamatoria e inmunitaria18. La principal limitación del trabajo de Arrieta et al.1 es que solo aporta asociaciones y no permite establecer causalidad, al tratarse de un estudio trasversal, pero la evidencia previa acumulada en estos 20 años y los estudios experimentales5,14,19 nos permiten interpretar indirectamente esta causalidad y avanzar hacia la consideración del ST2 circulante como un patógeno que ocasiona fibrosis e hipertrofia, con progresión del remodelado adverso y la disfunción del miocardio.
Uno de los retos pendientes en la medicina cardiovascular es trasladar la información de riesgo de los muchos biomarcadores identificados durante estos años en términos de su aplicación terapéutica. Recientemente hemos visto que al menos parte del beneficio de la inhibición de neprilisina reside en el aumento de los efectos favorables de los péptidos natriuréticos al evitar su eliminación. En el caso del ST2 circulante o soluble, su efecto patógeno se explicaría por la inhibición de la interacción cardioprotectora entre la IL-33 y el receptor de membrana ST2L, cuya señalización evita la hipertrofia, la inflamación y la fibrosis19,20. Son muchos los modelos experimentales que durante estos años han mostrado el efecto protector del ST2L en los cardiomiocitos y su interacción con la IL-3314,21,22 y al contrario, el efecto perjudicial de su deleción genética23. En este mismo modelo, la adición de la isoforma soluble sST2 conlleva la pérdida de la cardioprotección, por un secuestro inefecaz de IL-3324. Por otro lado, fármacos como los mineralocorticoides han mostrado que al menos parte de su beneficio vendría de una regulación favorable de la señalización del ST219,22.
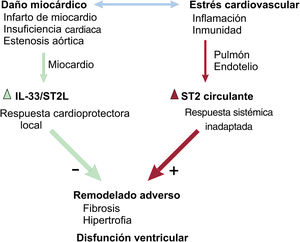
Por lo tanto, podemos afirmar que ante un daño cardiaco se producen 2 respuestas: una local cardioprotectora en el miocardio y otra sistémica, inflamatoria e inadaptativa que causaría la pérdida de esa respuesta local favorable (fig. 1). La siguiente pregunta sería cómo potenciar esta señalización cardioprotectora, y la evidencia acumulada indica que la respuesta es evitando la producción de la isoforma soluble o circulante del ST2, la misma que cuando está aumentada identifica una peor evolución en diferentes enfermedades cardiovasculares que conllevan un daño miocárdico, incluida la estenosis aórtica grave3,5,20. Nuestro grupo recientemente ha identificado elementos clave que regulan la expresión de esta isoforma soluble y circulante del ST225.
 y otra sistémica inadaptativa que conlleva aumento de ST2 soluble en la circulación que ocasionaría un bloqueo miocárdico de la señalización favorable, con más remodelado adverso, hipertrofia, fibrosis y disfunción cardiaca.")
Esquema de la interacción del ST2 y el sistema cardiovascular. En respuesta al daño cardiaco y el estrés cardiovascular, se produce una respuesta local cardioprotectora (ST2L-IL33) y otra sistémica inadaptativa que conlleva aumento de ST2 soluble en la circulación que ocasionaría un bloqueo miocárdico de la señalización favorable, con más remodelado adverso, hipertrofia, fibrosis y disfunción cardiaca.
Así, el trabajo de Arrieta et al.1 apoya el papel del ST2, no como un simple biomarcador de riesgo, sino como un patógeno cardiovascular que determina la aparición de disfunción y remodelado cardiaco, con hipertrofia y fibrosis miocárdica. En este contexto, la búsqueda de tratamientos capaces de disminuir su circulación y conseguir un efecto cardioprotector es un paso necesario.
FINANCIACIÓNA. Lax es investigador «Ramón y Cajal» en el Departamento de Medicina de la Universidad de Murcia (RYC2019-027635-I) y M.C. Asensio-López es investigadora «Juan de la Cierva» en el laboratorio de Fisiopatología Hematovascular del Centro Nacional de Investigaciones Cardiovasculares (FJC2020-042841-I). Se declara el apoyo de becas del Instituto de Salud Carlos III (PI19/00519; PI14/01637).
CONFLICTO DE INTERESESLos autores son inventores de una patente relacionada con la modulación del ST2 (PCT/EP2021/065521).