Las cardiopatías familiares se caracterizan por tener origen genético y la posibilidad de afectar a varios miembros de una misma familia. La cardiopatía familiar más frecuente es la miocardiopatía hipertrófica, con una prevalencia de 1:5001. En las últimas décadas se ha incrementado el conocimiento de la base molecular de estas enfermedades, y el análisis genético ha dado el salto a la práctica clínica2,3.
Presentamos el caso de una familia con miocardiopatía hipertrófica. El caso índice es una mujer de 31 años, diagnosticada en 1995 y tratada con propranolol 300 mg/día. Ingresó en nuestro servicio por primera vez en 2010 por síncopes de repetición. En el electrocardiograma había signos de hipertrofia ventricular izquierda. El ecocardiograma mostraba un septo interventricular de 27 mm, un gradiente máximo de 70 mmHg, movimiento sistólico anterior de la válvula mitral e insuficiencia mitral grado II. Tenía antecedentes de muerte súbita de familiares: dos tíos-abuelos maternos y un primo segundo, a los 20 años de edad mientras hacía deporte. Se decidió implantar un desfibrilador automático implantable, según las recomendaciones de las guías clínicas4.
La paciente acudió a la consulta de cardiopatías familiares solicitando consejo preconcepcional: tenía preembriones congelados en Estados Unidos, porque había intentado sin éxito la gestación de preembriones en una tercera persona. Estaba asintomática, y nunca había tenido descargas del desfibrilador automático implantable. Sus padres nunca habían sido valorados por un cardiólogo y su hermano no se había realizado un ecocardiograma desde 1995.
Tras explicar el tipo de herencia en esta enfermedad, autosómica dominante, se solicitó un ecocardiograma y una ergoespirometría, para estratificar el riesgo que conlleva el embarazo, y un test genético para dar el adecuado consejo genético. Presentó septo interventricular de 32 mm, gradiente de 60mmHg, insuficiencia mitral grado II y derrame pericárdico ligero. En la ergoespirometría alcanzó un consumo máximo de oxígeno de 26,1 ml/kg/min (el 73% del valor predicho), que no contraindica el embarazo. No obstante, se informó a la paciente del alto riesgo de complicaciones cardiacas. El test genético se realizó tras la obtención del consentimiento informado para los genes sarcoméricos más frecuentes (MYBPC3, MYH7, TNNT2, TNNI3, TPM1), y fue positivo para dos mutaciones en el gen MYBPC3 (NM_000257.2). La primera era de tipo missense, con un cambio de nucleótido c.13G>C;G5R, descrito previamente en la literatura5; la segunda variante era de tipo inserción: c.3066dupC;N1023fs+28X, que no se había descrito previamente, pero dado que ocasiona un cambio del marco de lectura, que origina un truncamiento de la proteína, se considera probablemente patogénica de la cardiopatía.
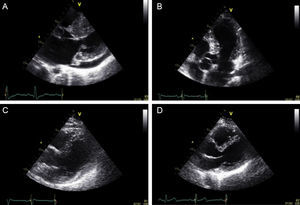
El hermano tenía 37 años, un electrocardiograma con ondas T picudas en precordiales izquierdas y un fenotipo característico de miocardiopatía hipertrófica: un septo interventricular de 19 mm, con presencia de fibrosis en la resonancia. El test genético fue positivo para una única mutación, c.3066dupC;N1023fs+28X. El padre tenía 63 años, hipertensión arterial, un electrocardiograma con repolarización precoz y un fenotipo dudoso de miocardiopatía hipertrófica por este antecedente: el septo interventricular era de 16 mm y la pared posterior, de 10 mm. El test genético fue positivo para la primera mutación (c.13G>C;G5R). La madre tenía 60 años, electrocardiograma y ecocardiograma normales (septo interventricular, 7 mm) (fig. 1). El test genético resultó positivo para la segunda mutación (c.3066dupC;N1023fs+28X) (fig. 2).


En resumen, el caso índice tenía dos mutaciones en MYBPC3 en alelos diferentes, porque cada una se había transmitido de un progenitor. Por lo tanto, la probabilidad de transmitir una de las mutaciones es del 100%, y todos los preembriones tendrían posibilidad de contraer la enfermedad. Por este motivo no se puede hacer diagnóstico preimplantacional, y desaconsejamos a la paciente realizar diagnóstico prenatal. El consejo reproductivo que se puede dar para evitar que sus descendientes sufran la enfermedad son la adopción y la gestación mediante fecundación in vitro con donación de ovocitos.
Se había planteado la posibilidad de realizar diagnóstico preimplantacional una vez conocida la mutación causante. Los requisitos son los siguientes:
- •
Identificar la mutación que causa la enfermedad.
- •
Ausencia de contraindicaciones para la gestación y el tratamiento hormonal de la fecundación in vitro.
- •
Posibilidad de realizar un test genético rápido en los preembriones.
- •
Cumplir las exigencias legales.
Nuestra legislación permite realizar el diagnóstico preimplantacional para «la detección de enfermedades hereditarias graves, de aparición precoz y no susceptibles de tratamiento curativo posnatal con arreglo a los conocimientos científicos actuales, con objeto de llevar a cabo la selección embrionaria de los preembriones no afectos»6. La realización del diagnóstico preimplantacional deberá comunicarse a la autoridad sanitaria correspondiente, que informará con periodicidad al menos semestral a la Comisión Nacional de Reproducción Humana Asistida. Sin embargo, en la ley no existe una lista de enfermedades hereditarias consideradas «graves», lo que origina una gran ambigüedad sobre las enfermedades en que se puede realizar el diagnóstico preimplantacional. Como cada vez será mayor el número de solicitudes de consejo reproductivo, sería recomendable crear una lista de enfermedades hereditarias bien definida dentro del grupo de trabajo de las miocardiopatías de la Sociedad Española de Cardiología, en colaboración con otras sociedades científicas.
FINANCIACIÓNEl presente trabajo se ha financiado parcialmente por la Red de Centros Cardiovasculares (RECAVA) apoyada por el Instituto de Salud Carlos III.