Fabry disease (FD) is a rare, progressive lysosomal storage disorder caused by a functional deficiency of the lysosomal α-galactosidase enzyme (α-gal A).1 It is an X-chromosome inheritance-linked disorder caused by pathogenic genetic variants in the GLA gene. These produce a functional enzyme deficiency, provoking intracellular accumulation of glycosphingolipids, predominantly globotriaosylceramide (lyso-Gb3) in lysosomal and nonlysosomal compartments of the skin, heart, kidney, brain, and other tissue cells, which contributes to the multisystemic manifestation of this disorder and early patient death.1,2
Fabry disease was first described among male patients with a severe form of the disease, a clinical phenotype now known as the classic form of the disease.1,2 These patients are usually carriers of nonsense or frameshift genetic variants that generate an absence of, or severely reduced, α-gal A activity, with symptom onset during infancy or adolescence, progressive failure in multiple organs and, finally, early death.1–3 However, a much larger group of patients are usually carriers of other genetic variants that do not cause such large changes in the protein structure (such as the majority of missense mutations) and give rise to variable levels of residual α-gal A activity, which might explain the late onset, less severe phenotypes encountered in the phenotypic expression of the disorder.3
Women are heterozygous for genetic variants in the GLA gene and manifest a heterogeneous clinical spectrum that varies between the absence of symptoms and a severity similar to that of male patients. Severity depends in part on the genetic variant and the pattern of X-chromosome inactivation, known as lyonization, which is a random process. Therefore, heterozygous patients who predominantly express a nonmutated GLA allele experience few or no symptoms, whereas patients who predominantly express a mutated GLA allele may experience a progression of the disorder similar to the masculine phenotypes, whether in a classic or late-onset form, depending on the underlying genetic variant in their family.4 Through this random inactivation of the mutated allele, in women α-galactosidase and lyso-Gb3 activity could show normal values.
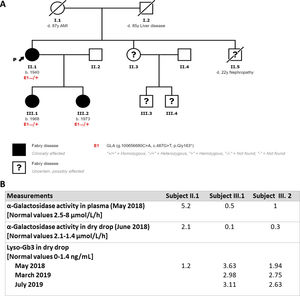
We present the case of a 78-year-old woman in follow-up for hypertrophic cardiomyopathy (HCM), with multiple supraventricular and ventricular arrhythmic episodes, and an implantable cardioverter-defibrillator for primary prevention. She also had stage 4A2 chronic kidney disease with multiple renal cysts and proteinuria, in addition to corneal deposits attributed to chronic treatment with amioderone. To complete the study, we performed an HCM genetic panel (ultra-sequence, 104 genes) detecting a pathogenic GLA genetic variant (p.Gly163*), which allowed a diagnosis of FD with cardiac, kidney and ocular (cornea verticillata) involvement. Enzyme activity and lyso-Gb3 were measured and were within normal ranges (figure 1B).

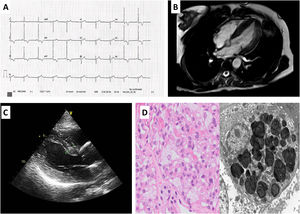
The familial study (figure 1A) found a brother who died at the age of 22 years due to a kidney condition, and a sister and 2 nephews with psychiatric disorders who did not wish to participate in the study. We assessed the index patient's 2 daughters, aged 52 and 47 years, who carry the same genetic variant and have FD. Neither of these 2 patients has children. The elder of the daughters has stage 2 A2 kidney involvement with significant proteinuria and mild left ventricular hypertrophy (figure 2B,C). The younger daughter had mild kidney involvement with microalbuminuria and slight ocular involvement. On physical examination, there were no skin lesions or acroparesthesia symptoms. Both patients showed reduced levels of enzymatic activity, and analyses also showed high levels of lyso-Gb3 in both (figure 1B). The kidney biopsy performed found lysosomal lipid storage consistent with the zebroid bodies characteristic of FD (figure 2D). In addition, the neurological and otorhinolaryngological studies found no abnormalities. We began enzyme replacement therapy for the 2 young patients with no complications or adverse effects, and without progression of kidney or heart involvement with a 2 year follow-up.

A: electrocardiogram of our index case's elder daughter showing a short PR interval, hypertrophy of the left ventricle and subepicardial ischemia. B and C: cardiovascular magnetic resonance and echocardiogram with hypertrophy of the left ventricle. D: optical microscope (left) hematoxylin and eosin stain glomerulus with enlarged podocytes due to cytoplasmic vacuolization. Electron microscope (right) shows abundant laminated cytoplasmic inclusions (myelin figures or zebroid bodies).
The genetic variant found in this family (p.Gly163*) has not been previously described, but it is a nonsense type mutation which, as a rule, is a pathogenic genetic variant because it produces a premature stop codon, giving rise to aberrant transcription or truncated peptides. Clinical information is available for at least 67 genetic variants of the same type clearly linked to FD. Some that are similar to our case have been included in a study of female patients with a diagnosis of FD,5 which observed that almost all of the women were symptomatic with findings of multisystemic disease.
Diagnosis of FD represents a challenge and it is often the cardiologist who suspects it in cases of HCM. FD is a disease with multiple manifestations that often delay diagnosis, requiring a multidisciplinary approach that allows us to identify these patients early and establish the best management and treatment. Clinical suspicion and imaging tests are essential in the differential diagnosis between HCM and FD, in addition to the utility of the genetic study. Our case is an example of how a genetic study with ultrasequencing is crucial in diagnosing cases among female patients with late onset and/or little cardiac involvement. The authors confirm that the written consent of the patients has been obtained for the submission and publication of this article, including the images and associated text. It has also been approved by the ethics committee.
FUNDINGNo funding related for this work.
AUTHORS’ CONTRIBUTIONSA. Robles-Mezcua has collected and analyzed the data as well as drafted the document. L. Morcillo-Hidalgo and M. Martín-Velázquez have contributed to the collection and analysis of data. M. León-Fradejas has carried out the pathological study of the biopsy samples and the corresponding report. J.M. García-Pinilla has reviewed the data collected and its analysis and revised the drafting of the final document.
CONFLICTS OF INTERESTNone declared.