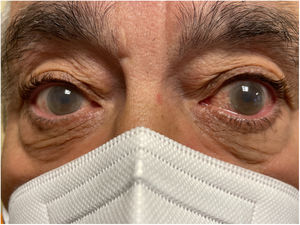

Primary hypoalphalipoproteinemia is an uncommon disorder characterized by the abnormal production of apolipoprotein (apo) A-I, resulting in extremely low levels of high-density lipoprotein cholesterol. The image shown is of a 60-year-old man, with consanguineous parents, with high-density lipoprotein cholesterol levels of 7mg/dL (40-60), triglycerides of 135mg/dL, total cholesterol of 119mg/dL, low-density lipoprotein cholesterol of 83mg/dL, lipoprotein A of 15.8mg/dL (< 30), and apoB of 99mg/dL (55-140). He was on treatment with simvastatin 10mg daily.
Examination findings of note included xanthelasma and complete corneal arcus with bilateral corneal opacities that did not affect visual acuity (figure 1). The patient gave written consent for his photographs to be taken and for the case to be published. The initial diagnostic suspicion was a lecithin-cholesterol acyltransferase deficiency, as corneal opacities are classically associated with this condition, the partial form of which is known as fish-eye disease. A genetic study was performed, which found a homozygous mutation in the apoA-1 gene associated with primary hypoalphalipoproteinemia (NM_000039.3(APOA1):c.67C>T (p.Gln23*)). For an accurate diagnosis, genetic testing is essential. Currently, management consists of controlling low-density lipoprotein cholesterol levels, and the prognosis is determined by premature onset of atherosclerotic disease.
FUNDING
No funding was received for the writing and publication of this work.
AUTHORS’ CONTRIBUTIONSJ. Costas Eimil: writing and design. P. Sánchez-Sobrino: review and approval.
CONFLICTS OF INTERESTThe authors declare no conflicts of interest.