Keywords
INTRODUCTION
Traditionally, the therapeutic options for patients with pulmonary arterial hypertension (PAH) associated with congenital shunts were limited to palliative measures, consisting of avoiding any factor that might destabilize the physiological equilibrium. Heart-lung transplantation was reserved only for a few highly selected patients.
Currently, based on the pathogenic and histologic similarity with different forms of pulmonary hypertension included within the PAH group according to the Venice 2003 classification, other forms of medical treatment are being tried with new drugs of proven benefit in different forms of PAH. Different drugs, such as synthetic prostacyclin and its analogues, phosphodiesterase inhibitors, and endothelin receptor antagonists, have demonstrated their usefulness in patients with idiopathic PAH. However, there was little scientific evidence of the efficacy of these drugs in patients with congenital heart disease with shunts and/or Eisenmenger syndrome (ES). Recently, prostacyclin analogues have been shown to improve functional capacity, oxygen saturation, and pulmonary hemodynamics in patients with PAH associated with congenital shunts.1 Given that the administration of prostacyclin is intravenous and prolonged therapy has often been associated with complications such as sepsis, orally administered drugs are preferred.
Bosentan, a dual endothelin receptor antagonist, has also been proposed as a pharmacological strategy in these patients. The Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5), designed to evaluate the effects of bosentan on systemic oxygen saturation, pulmonary and systemic hemodynamics, and exercise tolerance of patients with ES, confirmed the safety profile of the drug in these patients, who achieved significant improvements in hemodynamics and exercise tolerance, without any negative effects on systemic arterial oxygen saturation.2 However, the BREATHE-5 study only included patients with simple congenital heart disease with ES, which left unanswered questions such as whether bosentan was safe and effective in patients with complex congenital heart disease. Our objective was to assess the long-term effects of bosentan in patients with complex congenital heart disease and ES.
METHODS
Between September 2004 and June 2008, we treated 10 patients with complex congenital heart disease and ES with bosentan. All patients signed an informed consent prior to the study. ES was defined as a systemic-pulmonary predominantly right-to-left or two-way shunt due to increased pulmonary vascular resistance. All patients were aged over 18 years, had undergone a hemodynamic study to confirm diagnosis, and were in functional class 2-4 of the New York Heart Association (NYHA) classification. These 10 patients included 4 with truncus or pseudotruncus arteriosus, 2 with single ventricles, 2 with great artery transpositions, 1 with atrioventricular canal and Down syndrome, and 1 with ventricular septal defect with agenesia of the main branch pulmonary artery. Treatment with bosentan began on an outpatient basis at a dose of 62.5 mg/12 hours. After 1 month, if the drug was well tolerated, the dose was doubled to reach the optimal dose of 125 mg/12 hours.
Clinical follow-up of these patients was performed by monthly check-ups in our hospital unit. These check-ups included a medical history, physical examination, resting oxygen saturation, blood pressure, and full laboratory analysis with cell counts, transaminases, renal function, and uric acid. The clinical progression, drug tolerance, and NYHA functional class were assessed at each visit. In addition, before starting treatment and on successive visits (approximately once every 6 months), a 6-minute walk test was done as an objective assessment of the functional capacity, analyzing the distance covered and oxygen saturation at peak effort.
Continuous variables were expressed as mean (SD), and categorical ones as percentages. Given the small sample size, it was checked that each of the variables analyzed in the study followed a normal distribution using the Kolmogorov-Smirnov test. The study variables were compared using the paired Student t test. Statistical significance was established at P<.05.
RESULTS
The mean age of these patients at the start of treatment was 36 (10) years, with a predominance of women (70%). The treatment duration varied between 17 and 33 months (mean, 25 [8] months), and all patients reached the optimal bosentan dose without any abnormalities in liver enzymes or other indications of side effects. With regard to arterial oxygen saturation, there were no changes in values at rest or at peak effort: mean resting values at the beginning and end of treatment were 76% and 78%, respectively, while the peak effort values were 56% and 54%, respectively.
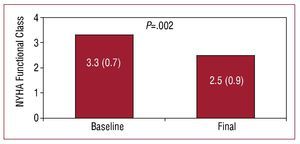
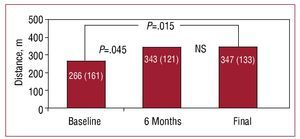
At baseline, 4 patients were in functional class 4, 5 in functional class 3, and 1 in functional class 2. All patients experienced a significant improvement in functional class, except for one female patient with truncus arteriosus, severe truncal valve regurgitation, and ventricular dysfunction, who remained in functional class 4 after 2 years of treatment at the optimal dose. This patient, after hemodynamic reassessment that found an elevated pulmonary wedge pressure, underwent successful heart-lung transplantation. The NYHA functional class at the end of follow-up in the remaining 9 patients changed from 3.3 (0.7) to 2.5 (0.9) (P=.002) (Figure 1). The distance covered in the 6-minute walk test increased from 266 (161) m to 347 (133) m (P=.015), with a net gain of 81 m during follow-up (Figure 2).

Figure 1.Changes in NYHA functional class during follow-up of patients with complex congenital heart disease and Eisenmenger syndrome in treatment with bosentan.

Figure 2.Changes in distance covered in the 6-minute walk test at baseline, 6 months, and at the end of follow-up in patients with complex congenital heart disease and Eisenmenger syndrome in treatment with bosentan.
DISCUSSION
There is currently no evidence-based treatment for patients with advanced PAH associated with congenital shunts, and so therapeutic recommendations are essentially based on expert opinion. The BREATHE-5 study has confirmed the safety profile and clinical and hemodynamic improvement associated with bosentan treatment in patients with ES, although only in patients with atrial and/or ventricular septal defects as the underlying congenital heart condition.
In our unit, we treated 10 patients with complex congenital heart disease and ES—all of whom were in NYHA functional class 2-4—with bosentan. Clinical follow-up in hospital, performed monthly, was done, including resting oxygen arterial saturation as a means of carefully monitoring for side effects, such as worsening of systemic hypoxemia due to widespread systemic vasodilation, and complete laboratory work-up with measurement of transaminases to check for liver function. Before starting treatment and every 6 months thereafter, the 6-minute walk test was done as an objective measure of functional capacity. This test is the most important one for determining the prognosis of idiopathic PAH3 and has been used in previous clinical trials to assess treatment response among patients with PAH associated with congenital shunting.4,5
With a long-term clinical follow-up (at least 17 months and a mean of 25 months), we evaluated the good tolerance towards bosentan in these patients, and all reached the optimum doses of 125 mg/12 hours without any liver enzyme abnormalities or other side effects. In accordance with previously published studies, bosentan administered to patients with congenital heart disease led to an improvement in exercise tolerance without negatively affecting systemic oxygen saturation. However, there was a significant variation in the clinical parameters assessed in the study: the NYHA functional class and distance covered in the 6-minute walk test. The only patient who withdrew from our study had been receiving treatment for 2 years. She was showing no improvement and remained in functional class 4. We are of the opinion that the main cause for the lack of response to treatment might have been the severe regurgitation of the truncal valve that led to severe ventricular dysfunction.
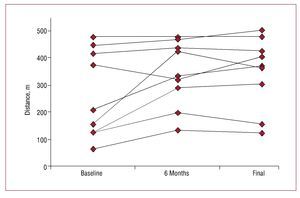
As in our study, Diller et al5 reported an improvement in functional class and distance covered in the 6-minute walk test with long-term bosentan administration in patients with PAH associated with congenital heart disease. Their patients were more heterogenous and included 6 patients with complex congenital heart disease, although the authors did not analyze the results for these patients separately. The improvement observed in our patients was observed through a significant increase in the distance covered in the 6-minute walk test performed every 6 months in the follow-up visits: 343 (121) m (P=.045) which, unlike recently published studies, is maintained during follow-up lasting more than 2 years5 (Figures 2 and 3).

Figure 3.Individual changes in distance covered in the 6-minute walk test at baseline, 6 months, and at the end of follow-up in patients with complex congenital heart disease and Eisenmenger syndrome in treatment with bosentan.
This study is subject to a series of limitations, such as the lack of a control group and the small sample size, in view of the low prevalence of this condition. With regard to the study design, the fact that we did not use invasive methods to monitor hemodynamic parameters was justified in that many pulmonary hypertension units have stopped doing right cardiac catheterization to assess treatment response, and the 6-minute walk test has become accepted as the most important prognostic test in PAH.6
In conclusion, in our experience, bosentan has extended its indications as it has been shown to be clinically effective in patients with ES and complex congenital heart defects, and clinical benefit is maintained over long treatment periods.
Correspondencia: Dr. J.M. Oliver Ruiz.
Unidad de Cardiopatías Congénitas del Adulto. Hospital Universitario La Paz.
P.o de la Castellana, 261. 28046 Madrid. España. E-mail: joliver.hulp@salud.madrid.org
Received March 8, 2008.
Accepted for publication November 24, 2008.