La rehabilitación cardiovascular es una estrategia terapéutica necesaria, segura y con evidencia científica para tratar la cardiopatía isquémica y reducir su morbimortalidad. No obstante, está subutilizada en todo el mundo. Esta revisión se centra en la potencialidad de esta estrategia para generar cardioprotección y, particularmente, en la capacidad del ejercicio físico para inducir un fenotipo cardioprotector contra el daño generado por la isquemia-reperfusión. Se abordan los principales mecanismos moleculares del daño y de la protección, incluyendo el precondicionamiento isquémico inducido por el ejercicio. También se tratan adaptaciones cardioprotectoras que el ejercicio físico genera en el tejido vascular y el sistema autónomo.
adenosintrifosfato.
isquemia-reperfusión.
precondicionamiento isquémico.
poros de permeabilidad transitoria.
rehabilitación cardiovascular.
respecies reactivas del oxígeno.
Los programas de rehabilitación cardiovascular (RC) se definen como un conjunto de medidas terapéuticas para el cuidado integral de los pacientes con enfermedad cardiovascular; su recomendación se considera segura, útil y efectiva (clase I-A), especialmente en sujetos con enfermedad coronaria e insuficiencia cardiaca crónica1,2.
Hay evidencia científica del impacto favorable de los programas de RC en el proceso fisiopatológico de estas enfermedades y de la reducción de la mortalidad entre un 20 y un 25%. No obstante, están subutilizados en todo el mundo. En Estados Unidos, menos del 30% de los pacientes elegibles son referidos a estos programas1-5.
A partir de estas reflexiones de beneficio y subutilización contradictoria de estos programas, decidimos que es necesario apoyar el nivel de evidencia del beneficio que producen. Por lo tanto, brindamos en nuestra revisión justificaciones novedosas a nivel molecular sobre la potencialidad de la RC para generar cardioprotección endógena, centrándonos en la capacidad del ejercicio físico para inducir un fenotipo cardioprotector contra el daño generado por la isquemia-reperfusión (IR) y en las adaptaciones cardioprotectoras que el ejercicio físico genera en el tejido vascular y el sistema autónomo.
Para una mejor comprensión de los efectos cardioprotectores del ejercicio físico contra el daño por IR, creemos prudente dejar claro cuáles son los cambios celulares que se producen en esta circunstancia.
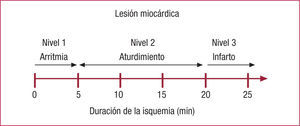
¿QUÉ OCURRE DURANTE LA ISQUEMIA-REPERFUSIÓN?La magnitud del daño de la IR está determinada por la duración de la isquemia. Los niveles del daño van desde el generado por una breve agresión que resulta en un daño no permanente, hasta los generados por eventos duraderos que pueden llevar a la disfunción eléctrica cardiaca y la muerte celular. Las isquemias que duran hasta 5min (fig. 1) no causan disfunción contráctil ventricular o muerte celular, pero pueden producir arritmias. Las isquemias que duran hasta 20min producen disfunción contráctil ventricular pero no muerte celular, y las que duran más de 20min son las que producen muerte celular. El daño en la reperfusión ocurre durante sus primeros 60s, y su magnitud está determinada por la duración de la isquemia6.

En la IR hay depleción de energía, acumulación de metabolitos, estrés oxidativo y sobrecarga de calcio; la extensión del daño depende de la magnitud de estos cambios7.
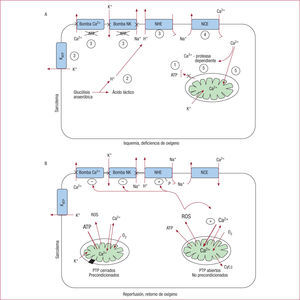
En la isquemia (fig. 2A), con la pérdida de oxígeno viene un inmediato cese de la producción de adenosintrifosfato (ATP) mitocondrial, lo que dispara un incremento de la glucolisis anaeróbica para tratar de compensar la necesidad de energía. Como resultado, se incrementan las concentraciones de iones de hidrógeno y ácido láctico en el citosol, lo que genera una inhibición de la glucolisis anaeróbica con la depleción total de energía. Las concentraciones intracelulares de Na+ se incrementan porque el pH intracelular bajo activa el intercambiador sodio-hidrógeno (NHE) en el sarcolema y porque, junto con la falta de energía, se inhibe en el sarcolema la bomba Na+/K+dependiente de ATP. En esta situación también se inhibe la bomba de Ca2+ del sarcolema y del retículo sarcoplásmico. La depleción de ATP también deriva en la apertura de los canales de K+ dependientes de ATP del sarcolema y las mitocondrias. La apertura de estos canales se considera un mecanismo de defensa para reducir las especies reactivas de oxígeno (ROS) generadas durante la reperfusión7,8.
 PTP mitocondriales. El K+está entrando en la mitocondria por la apertura de los canales de K+ sensibles a
PTP mitocondriales. El K+está entrando en la mitocondria por la apertura de los canales de K+ sensibles a Algunos eventos que ocurren durante la isquemia (A) y durante la reperfusión (B). Los números en el panel A representan la secuencia de eventos. En el panel B se visualiza cómo la mitocondria puede responder si está precondicionada o no ante una forma inespecífica de precondicionamiento y el papel de los PTP mitocondriales. El K+está entrando en la mitocondria por la apertura de los canales de K+ sensibles a ATP mitocondriales. ATP: adenosintrifosfato; Cyt.c: citocromo C; NCE: intercambiador Na+/Ca+; NHE: intercambiador Na+/H+; PTP: poros de permeabilidad transitoria; ROS: especies reactivas de oxígeno.
Con el retorno del oxígeno durante la reperfusión (fig. 2B), la mitocondria genera una gran cantidad de ROS. La sobrecarga de Na+ intracelular continúa durante la fase inicial de la reperfusión, porque los NHE son activados por ROS y, como aún hay escasez de ATP, hay una demora en la reactivación de las bombas de Na+/K+ y Ca2+ dependientes de ATP en el sarcolema. La alta concentración de Na+ hace que el intercambiador Na+/Ca2+ (NCE) actúe de manera inversa, produciendo una sobrecarga de Ca2+ en el citosol y la mitocondria. La sobrecarga de Ca2+ deriva en una mayor producción de ROS y la activación de proteasas, como las calpaínas, que generan degradación de proteínas, entre ellas la ATPasa-Ca2+ de retículo sarcoplásmico (SERCA2a). Esta situación conduce a una disfunción de las proteínas contráctiles8,9. La sobrecarga de Ca2+ y ROS produce la apertura de los poros de permeabilidad transitoria (PTP) mitocondriales. La apertura de estos poros inespecíficos de gran conductancia en la membrana mitocondrial produce la liberación de citocromo c al citosol, lo que induce la activación de las caspasas10,11. Si la lesión por la IR es grave, los poros permanecen abiertos y la célula muere vía necrosis; si es moderada, vía apoptosis, y si es ligera, se produce la recuperación10,11.
¿Cómo protege el ejercicio físico del daño por isquemiareperfusión?El ejercicio físico per se o mediante la inducción de precondicionamiento isquémico (PI) genera adaptaciones moleculares de magnitud citoprotectora, con la expresión y modulación genética de un fenotipo cardioprotector que mitiga casi todos los mecanismos fisiopatológicos del daño por IR12-21.
Según resumen Powers et al22, entre los mecanismos que el ejercicio induce para generar cardioprotección contra el daño por IR, parecen no ser esenciales la formación de colaterales23, el incremento de proteínas de choque térmico24,25, el incremento en la expresión de la ciclooxigenasa 226 y el alto nivel de proteínas de estrés del retículo endoplásmico27. Por lo tanto, nos centraremos en los mecanismos que han resultado ser condición sine qua non para esta protección, como el incremento de la protección antioxidante, los cambios en el metabolismo y la expresión proteica mitocondrial, el incremento en la expresión y la apertura de los canales de K+ATP mitocondriales y del sarcolema y la atenuación de la activación de las calpaínas por la IR.
Protección antioxidanteLa producción de ROS, la acumulación de iones de hidrógeno y la generación de especies reactivas de nitrógeno desempeñan un papel muy importante en el complejo fisiopatológico del daño por IR28. La mitocondria es la fuente primaria en la producción de ROS durante la IR29. En un modelo animal, se ha comprobado que el ejercicio físico protege al cardiomiocito del estrés oxidativo que genera la IR y a sus mitocondrias del daño estructural asociado a esta noxa30.
Hay consenso en que el incremento de la protección antioxidante generada por el ejercicio se debe a un incremento en la expresión de las enzimas manganeso y cobre-cinc superóxido dismutasas (MnSOD y Cu-ZnSOD), la glutatión peroxidasa (GPX) y las catalasas (CAT)31,32. De las SOD, la más importante es la MnSOD, y sólo se incrementa con ejercicios de alta intensidad. Estudios recientes han demostrado que la MnSOD es esencial para la protección completa contra el infarto miocárdico y la taquicardia ventricular inducidos por IR33.
Cambios en el metabolismo y la expresión proteica mitocondrialLas mitocondrias cardiacas del subsarcolema e intermiofibrilares contribuyen en la protección inducida por el ejercicio físico contra el daño por IR12,14 mediante algunas de las siguientes vías: la reducción en la producción de ROS por las mitocondrias cardiacas34,35; las mitocondrias son más resistentes a la apertura de los PTP mitocondriales inducida por el Ca2+, lo que evita que se desencadenen potentes mecanismos apoptóticos34; la inducción de una reducción en la expresión mitocondrial de la monoaminooxidasa A, que parece ser una fuente importante de estrés oxidativo36,37.
Papel de los canales de K+ATP mitocondriales y del sarcolemaLa activación de los canales de K+ATP mitocondriales atenúa la sobrecarga de Ca2+ y, por lo tanto, previene la apertura de los PTP mitocondriales y el daño que esto ocasiona, aunque algunos investigadores desestiman que el ejercicio induzca este mecanismo38. El mecanismo protector del ejercicio se asocia más con la apertura de los canales K+ATP del sarcolema, que permiten una aceleración de la repolarización por incremento en la salida de K2+ y un acortamiento de la duración del potencial de acción. Como resultado de este acortamiento, se previene la sobrecarga de Ca2+ por la reducción del tiempo de apertura de los canales de Ca2+ tipo L39.
Atenuación de la activación de la calpaína inducida por isquemiareperfusiónLa calpaína es una proteasa dependiente de Ca2+ con dos isoformas (calpaína I y calpaína II), que se activan por una exposición prolongada a niveles altos de Ca2+ en el citosol y contribuyen al daño por IR40. French et al41 mostraron que la inhibición de la calpaína atenuó la disfunción contráctil inducida por IR y que el ejercicio redujo la activación de la calpaína asociada a IR, porque mejoró la protección antioxidativa con incremento de la MnSOD y las CAT y previno la degradación de la SERCA2a y la fosfolamban, mecanismos adaptativos que generan una reducción de la concentración de Ca2+ en el citosol y, por lo tanto, atenuación de la activación de la calpaína, que depende de Ca2+.
Umbral y duración de la sesiones de ejercicio para lograr cardioprotecciónPara lograr los efectos cardioprotectores mediante el ejercicio físico, la duración de una sesión de ejercicio debe ser de al menos 60min, con una intensidad en que la frecuencia cardiaca de entrenamiento sea el 75% del consumo pico de oxígeno. Hay evidencias de que la cardioprotección contra el aturdimiento miocárdico inducida por el ejercicio persiste a los 9 días de haber cesado el entrenamiento y se pierde totalmente a los 18 días9,36.
Inducción de precondicionamiento isquémico por ejercicio físico. Cardioprotección y potencial aplicación en la rehabilitación cardiovascularDesde hace más de 5 años, la American Heart Association (AHA) reconoce el PI inducido por el ejercicio como un mecanismo cardioprotector potente, de utilidad dentro de los programas de RC en pacientes con enfermedad coronaria avanzada, en quienes la realización de ejercicios a umbrales isquémicos incrementa la tolerabilidad miocárdica para enfrentarse a ulteriores situaciones de estrés isquémico prolongado, con la consiguiente reducción del daño miocárdico y el riesgo de sufrir taquiarritmias ventriculares letales42.
A partir de evidencias científicas, se considera segura la implementación de esta modalidad de RC, que debe ser individualizada y con vigilancia electrocardiográfica estricta, para que, durante una sesión de entrenamiento de 60min, el paciente alcance de forma intermitente el umbral isquémico y lo mantenga durante periodos>90s y<5min19,20,43-45.
Según algunos autores46, los mecanismos moleculares del PI para generar un fenotipo citoprotector son diversos: los que dependen de la modalidad —si es primera o segunda ventana de protección— o de la naturaleza de la isquemia precondicionante. Entre estos mecanismos se incluyen principalmente la apertura de los canales de K+del sarcolema y mitocondriales, el incremento en la actividad y expresión de la MnSOD, de la óxido nítrico sintasa endotelial (eNOS) y la inhibición de la apertura de los PTP mitocondriales durante la reperfusión16-18,21,36.
Efectos vasculares del ejercicio físico que se relacionan con cardioprotecciónEl ejercicio físico, per se o con la inducción de PI, ejerce efectos favorables en el sistema vascular que generan cardioprotección. Hambrecht et al47 realizaron el primer estudio en humanos que evaluó el efecto de un programa de ejercicio físico en la función endotelial coronaria, y encontraron que un programa de 4 semanas con ejercicios aeróbicos mejoró la función coronaria y que ese efecto se perdió parcialmente después de 5 meses de haber designado a los pacientes a un programa de menor intensidad en sus casas.
El estrés de cizallamiento laminar generado por el ejercicio produce estímulos mecánicos en el citoesqueleto endotelial que se transforman en vías de señalización bioquímicas a nivel molecular que promueven una mejor función endotelial48. Entre estas señalizaciones se incluyen la fosforilación y activación de la eNOS con el incremento en la producción de óxido nítrico (NO), el incremento en la expresión y activación de las enzimas antioxidantes SOD, CAT y GPX que, junto con la reducción en la expresión de la oxidasa del NADPH y de los receptores AT1 de la angiotensina II, producen una reducción de ROS y de la vasoconstricción mediada por la angiotensina II48-50.
Está en debate si el ejercicio físico per se o sólo cuando induce isquemia genera incremento de las células progenitoras endoteliales (CPE) en la médula ósea mediante la estimulación del factor de crecimiento endotelial, el factor de crecimiento placentario y el sistema de metaloproteinasas. Este incremento de las CPE permite mantener, en situaciones críticas, la capa de células endoteliales51-55.
Efectos del ejercicio físico en el sistema autónomo que se relacionan con cardioprotecciónEs ampliamente conocido que un programa de entrenamiento físico produce evolutivamente una reducción del incremento de la frecuencia cardiaca y del consumo miocárdico de oxígeno (MVO2) para una misma carga de trabajo. En los pacientes con enfermedad coronaria, estos cambios de la frecuencia cardiaca tienen un efecto favorable en el umbral isquémico, son cardioprotectores y mejoran la calidad de vida9.
Actualmente ha cobrado gran interés la capacidad del infarto de miocardio (IM) para generar una remodelación de la regulación autonómica cardiaca, con inestabilidad eléctrica y propensión a sufrir taquiarritmias ventriculares malignas, y la capacidad del ejercicio físico para generar un remodelado inverso y reducir tal propensión56.
El IM produce una remodelación de la regulación del sistema autónomo sobre el sistema cardiovascular, al reducir la regulación parasimpática y alterar la relación de los adrenorreceptores beta miocárdicos, incrementando la expresión y la actividad de los adrenorreceptores beta 2, mientras que los beta 1 permanecen constantes. Esto deriva en una desregulación del Ca2+ con sobrecarga intracelular y potencialidad para desarrollar arritmias malignas57,58. Por lo tanto, las intervenciones que aumenten la actividad parasimpática y reduzcan la actividad adrenérgica cardiaca podrían también proteger contra la fibrilación ventricular.
Hay suficiente evidencia científica en modelos animales y humanos de la capacidad de un programa de ejercicio físico tras un IM para producir una remodelación inversa en la regulación del sistema autónomo sobre el sistema cardiovascular y proteger contra la fibrilación ventricular59-64.
CONCLUSIONESSe ha tratado en nuestra revisión una gran cantidad de evidencias científicas, a nivel molecular, del efecto cardioprotector del ejercicio físico y de su beneficio para mejorar la morbimortalidad de las enfermedades cardiovasculares. Estos datos indican la necesidad de su empleo como una estrategia terapéutica óptima para tratar las enfermedades cardiovasculares y generar salud.
«El hombre que no tiene tiempo para hacer ejercicio, tendrá tiempo para enfermarse.»Sócrates.
CONFLICTO DE INTERESESNinguno.