En estudios aleatorizados amplios se ha establecido que un control intensivo temprano de la glucemia reduce el riesgo de complicaciones diabéticas, tanto microvasculares como macrovasculares. Sin embargo, los datos epidemiológicos y prospectivos respaldan la existencia de una influencia a largo plazo del control metabólico inicial sobre la evolución clínica posterior. A este fenómeno se le ha denominado recientemente “memoria metabólica”. Los posibles mecanismos para la propagación de esta “memoria” son la glicación no enzimática de proteínas y lípidos celulares, y el exceso de especies moleculares de nitrógeno y oxígeno reactivas celulares, en especial las originadas en las proteínas mitocondriales glicadas, que tal vez actúen de manera concertada entre sí para mantener las señales de estrés. Además, la aparición de esta “memoria metabólica” sugiere la necesidad de un tratamiento enérgico muy temprano destinado a “normalizar” el control metabólico y de la adición de fármacos que reduzcan las especies moleculares reactivas celulares y la glicación, además de normalizar las concentraciones de glucosa de los pacientes, para reducir al mínimo las complicaciones diabéticas a largo plazo.
Palabras clave
La diabetes constituye un problema de salud pública grave y creciente que provoca una reducción de la esperanza de vida y un aumento de la morbilidad debida a las complicaciones específicas de la propia enfermedad. El dato que caracteriza a la diabetes es la hiperglucemia, un factor de estrés que puede controlarse clínicamente mediante la administración exógena de insulina o mediante fármacos que aumentan la secreción de insulina, reducen la liberación de glucosa procedente del hígado, aumentan el uso de glucosa en el músculo esquelético y la grasa, retrasan la absorción de la glucosa procedente de los alimentos y, en productos introducidos muy recientemente, actúan a través del sistema de la incretina1. Estos avances, junto con la mejora de la monitorización de la glucosa y los mejores marcadores del control glucémico, han conducido a un control mucho más estricto de la hiperglucemia. A pesar de estos progresos terapéuticos, las complicaciones vasculares debilitantes se continúan dando en la mayoría de los pacientes diabéticos.
En el Diabetes Complications and Control Trial (DCCT), se aplicó a pacientes con diabetes tipo 1 un pauta de tratamiento estándar o de tratamiento intensivo para normalizar sus valores de glucemia. Dada la intensa reducción de la progresión de las complicaciones microvasculares observada en los pacientes con un control estricto de la glucosa, el DCCT se dio por finalizado tras una media de 6,5 años y todos los pacientes pasaron a recibir el tratamiento intensivo2. Es de destacar que, en el ensayo Epidemiology of Diabetes Interventions and Complications (EDIC), un estudio de seguimiento del DCCT, los pacientes que habían recibido la pauta de tratamiento estándar durante el DCCT continuaban presentando una incidencia de complicaciones superior a la de los pacientes que habían recibido un tratamiento intensivo durante todo el ensayo, varios años después de haber pasado al tratamiento intensivo3,4. Además, los datos recientes del EDIC sugieren también que la influencia del control glucémico temprano sobre la progresión hasta presentar episodios macrovasculares puede hacerse más evidente con un seguimiento más prolongado5,6.
Los datos del United Kingdom Prospective Diabetes Study (UKPDS) parecen coincidir con ello. Concretamente, los individuos que presentaban valores de glucosa plasmática en ayunas (GPA) más bajos en el momento del diagnóstico sufrieron menos complicaciones vasculares y menos resultados clínicos adversos a lo largo del tiempo que los individuos con valores de GPA más altos, a pesar de tener índices de aumento de la glucemia similares7, lo cual sugiere que el control metabólico temprano ejerce unos efectos beneficiosos mantenidos incluso en la diabetes tipo 2.
El conjunto de estas observaciones respalda el concepto de que el entorno glucémico inicial es recordado, y los autores del DCCT/EDIC se han referido a este fenómeno como una "memoria metabólica"6.
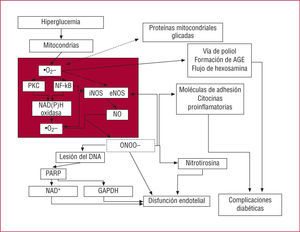
FUNDAMENTO MOLECULAR DE LA "MEMORIA METABÓLICA": POSIBLE VÍNCULO ENTRE EL ESTRÉS OXIDATIVO Y LA GLICACIÓN NO ENZIMÁTICAPapel del estrés oxidativo en las complicaciones diabéticasBrownlee ha señalado recientemente la presencia de un exceso de aniones superóxido (•O2-), una especie molecular reactiva, en las mitocondrias de las células endoteliales, en respuesta a la hiperglucemia, con la formación de complicaciones diabéticas8. Aun cuando el aumento de generación de •O2- en la hiperglucemia sea un proceso clave en la activación de otras vías que intervienen en la patogenia de las complicaciones diabéticas8, representa tan sólo un primer paso en la producción del estrés oxidativo celular global y la consiguiente lesión vascular a que éste conduce. La hiperglucemia favorece también, a través de la activación del NF-kβ, un aumento de la expresión de NAD(P)H y de iNOS9, que sería de prever que provo-cara un exceso de NO y de •O2-. Se cree que el NO contribuye a producir la disfunción endotelial de dos formas distintas. En primer lugar, el •O2- puede reac-cionar también directamente con el NO, atenuándolo, con lo que se reduce la eficacia de un potente sistema vasodilatador derivado del endotelio que participa en la homeostasis general de los vasos sanguíneos10, y la evidencia existente sugiere que durante la hiperglucemia se produce una disminución de la disponibilidad de NO11. En segundo lugar, como se ha mencionado antes, la sobreproducción de •O2-, cuando se acompaña de un aumento de la generación de NO, favorece la formación del oxidante potente ONOO, y se ha des-crito una sobreproducción de •O2- y NO en respuesta a la hiperglucemia12. Se ha comprobado que un aducto proteico estable, la 3-nitrotirosina (3-NY), es un mar-cador del ONOO-13 y del •NO214 y que puede determi-narse con facilidad mediante ELISA o western blot. La posibilidad de que la diabetes se asocie a un aumento de la formación de ONOO- está respaldada por la reciente detección de un aumento de las concentra-ciones plasmáticas de nitrotirosina en los pacientes con diabetes tipo 215. Hay varios elementos de evidencia que respaldan un papel directo de la hipoglucemia para favorecer este fenómeno. Se detecta la formación de 3-NY en la pared arterial de los monos durante la hiperglucemia16, en el plasma de individuos sanos durante un clamp hiperglucémico17 y en pacientes diabé-ticos durante un aumento de la hiperglucemia pospran-dial18. La hiperglucemia se acompaña también de un depósito de 3-NY en el corazón de rata perfundido en funcionamiento19, y es razonable pensar que esté rela-cionado con el desequilibrio en la producción de NO y •O2- a través de la sobreexpresión de iNOS19 y a través de los muchos orígenes de •O2- que se han descrito antes. La formación de 3-NY se asocia también a la aparición de una disfunción endotelial tanto en los individuos sanos17 como en las coronarias de corazones de rata perfundidos19. Es interesante señalar que, en clínica, se ha observado que la 3-NY es un factor predictivo independiente para la enfermedad vascular20. Todas las vías que se han descrito se resumen en la figura 1.

La hiperglucemia intracelular induce una sobreproducción de superóxido a nivel mitocondrial. Esto constituye el primer proceso crucial en la activación de todas las demás vías que intervienen en la patogenia de las complicaciones diabéticas, como el flujo de la vía de poliol, el aumento de la formación de AGE, la activación de la proteincinasa C y el NFkB y el aumento del flujo en la vía de la hexosamina.
Las proteínas mitocondriales sufren una glicación en la hiperglucemia y este efecto induce una superproducción de aniones superóxido en las mitocondrias. En este caso, a pesar de que la glucemia se reduzca o se normalice, las mitocondrias glicadas continúan sobreproduciendo superóxido, con lo que activan las mismas vías que intervienen en la generación de las complicaciones diabéticas. Esta hipótesis puede contribuir a explicar la aparición de la denominada "memoria metabólica".
De los datos presentados parece deducirse claramente que tanto el estrés oxidativo como el nitrosactivo desempeñan un papel clave en el desarrollo de las complicaciones diabéticas, tanto microvasculares como macrovasculares. Sin embargo, si el exceso de especies moleculares reactivas desempeña un papel central en el desarrollo de las complicaciones diabéticas relacionadas con la hiperglucemia, ¿podría explicar dicho exceso la persistencia del riesgo de complicaciones aun cuando se haya reducido o normalizado la hiperglucemia?
Hace varios años hubo una descripción preliminar de una "memoria hiperglucémica" para una hiperproducción de fibronectina y colágeno en las células endoteliales, que persistía tras la normalización de la glucosa21. Utilizando el mismo diseño, 14 días de cultivo con glucosa elevada, seguidos de 7 días de cultivo con glucosa normal, los datos preliminares indican que, en las células endoteliales, persiste una sobreproducción de radicales libres tras la normalización de la glucosa, y que ello se acompaña de una prolongación de la inducción de PKC-β, NAD(P)H oxidasa, Bax, colágeno y fibronectina, además de 3-NY22, lo cual sugiere que el estrés oxidativo puede intervenir en el efecto de "memoria metabólica".
Glicación de proteínas mitocondriales, estrés oxidativo y "memoria metabólica"Se ha sugerido la sobreproducción mitocondrial de •O2- en la hiperglucemia como "hipótesis unificadora" para explicar el desarrollo de complicaciones diabéticas8. Parece razonable pensar, pues, que las mitocondrias desempeñen también un papel importante en la propagación de la "memoria metabólica".
Se cree que la hiperglucemia crónica altera la función mitocondrial a través de la glicación de las proteínas mitocondriales23. Las concentraciones de metilglioxal (MGO), un producto α-dicarbonilo derivado de la glucólisis que es altamente reactivo, están aumentadas en la diabetes24. El MGO reacciona con facilidad con la arginina, la lisina y los grupos sulfihidrilo de las proteínas25, además de los ácidos nucleicos26, induciendo la formación de diversos AGE estructuralmente identificados, tanto en las células diana como en el plasma27. El MGO tiene un efecto inhibidor sobre la respiración mitocondrial, y las modificaciones inducidas por el MGO tienen efectos específicos en determinadas proteínas mitocondriales28. Estas premisas son importantes puesto que un estudio reciente ha descrito, por primera vez, una relación directa entre la formación de AGE intracelulares a partir de las proteínas mitocondriales, la disminución de la función mitocondrial y el exceso de formación de especies moleculares reactivas29. Así pues, las proteínas de la cadena respiratoria mitocondrial que sufrían una glicación eran propensas a producir más •O2-, independientemente del grado de hiperglucemia existente.
La formación de AGE es un fenómeno prolongado. En el DCCT, se examinó la formación de AGE en 215 pacientes a los que se practicó una biopsia cutánea 1 año antes del cierre del ensayo30. En comparación con el tratamiento convencional, el tratamiento intensivo se asoció a unas concentraciones de AGE significativamente inferiores. La aparición de retinopatía, nefropatía y neuropatía30 presentó una asociación significativa con los valores de AGE, y en el EDIC se ha observado que el aumento de las concentraciones de AGE en la piel se asocia de manera significativa a las complicaciones microvasculares31. Además, parece razonable pensar que los AGE puedan explicar también los resultados que indican un aumento de la incidencia de complicaciones cardiovasculares en el EDIC6, teniendo en cuenta que se ha observado una asociación de los AGE con la ECV incluso en las mujeres no diabéticas32.
Lo realmente importante es la evidencia clínica que indica que la inclinación de las proteínas, y en especial del colágeno, a la glicación es independiente de la concentración de glucosa existente en ese momento27. Se ha propuesto también que la glicación del colágeno extracelular puede ser un marcador de la glicación de proteínas intra celulares y un factor predictivo de la lesión de órganos diana27. Aunque la HbA1c glicada puede ser desglicada en parte enzimáticamente33, no se ha observado todavía una reacción de este tipo para los AGE incorporados al colágeno. Así pues, parece que la formación de AGE de colágeno es un fenómeno irreversible.
En resumen, la glicación de las proteínas mitocondriales puede formar parte de la explicación del fenómeno de la "memoria metabólica". En las mitocondrias glicadas hay una sobreproducción de radicales libres, independientemente de la glucemia real, que mantiene la activación de las vías involucradas en la patogenia de las complicaciones diabéticas. En otras palabras, puede plantearse la hipótesis de que en la "memoria metabólica" la cascada de procesos sea la misma que la propuesta por Brownlee8 —el origen del •O2- continúan siendo las mitocondrias— pero que, además, la producción de especies moleculares reactivas no está relacionada con la presencia de hiperglucemia y depende del nivel de glicación de las proteínas mitocondriales. Esta hipótesis se describe en la figura 1.
"MEMORIA METABÓLICA"Y DISFUNCIÓN ENDOTELIAL: TRASCENDENCIA PARA EL RIESGO CARDIOVASCULAR EN LA DIABETESLa diabetes mellitus se asocia a un aumento de la incidencia de enfermedades macrovasculares. La enfermedad macrovascular acelerada se debe en parte a un aumento de la incidencia de los factores de riesgo clásicos, como hipertensión y dislipidemia34. Sin embargo, la evidencia reciente sugiere que la hiperglucemia desempeña también un papel importante6.
El endotelio es un órgano importante en el desarrollo de la enfermedad cardiovascular incluso en la diabetes35. Todos los factores de riesgo que intervienen en la patogenia de la enfermedad cardiovascular, como la dislipidemia y la hipertensión, pueden inducir una disfunción endotelial, y se ha puesto claramente de manifiesto que ésta predice un futuro episodio cardiovascular35.
La presencia de una disfunción endotelial se ha descrito a menudo en la diabetes35. Sin embargo, aunque diversos estudios han puesto de manifiesto que la hiperglucemia induce una disfunción endotelial tanto en individuos diabéticos como en no diabéticos36,37, no disponemos todavía de una demostración clara de que el control de la hipertensión permita restablecer/normalizar la función endotelial. Concretamente, en pacientes con diabetes tipo 1, se ha descrito que la disfunción endotelial aparece a pesar de que se alcance una normoglucemia38,39. Además, hay varios estudios que indican que la hiperglucemia induce una disfunción endotelial a través de la generación de un estrés oxidativo, que se ha sugerido que es un factor clave en la generación de las complicaciones diabéticas, tanto microvasculares como macrovasculares8.
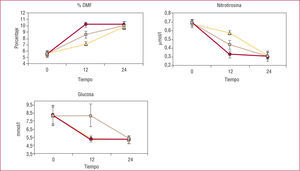
En un reciente estudio, se incluyó a 36 pacientes con diabetes tipo 1 y 12 individuos de control. Se dividió a los pacientes diabéticos en tres grupos40. El primero de ellos fue tratado durante 24h con insulina, y alcanzó una casi-normalización de la glucemia. A las 12h de este tratamiento, se añadió vitamina C durante las 12h restantes. El segundo grupo fue tratado durante 24h con vitamina C. A las 12h de este tratamiento se inició la administración de insulina, y se alcanzó una casi-normalización de la glucemia durante las 12h restantes. El tercer grupo fue tratado durante 24h con vitamina C e insulina, y alcanzó una casi-normalización de la glucemia. Ni la normalización de la glucemia ni el tratamiento con vitamina C sola lograron normalizar la disfunción endotelial o el estrés oxidativo. La combinación de insulina y vitamina C normalizó la disfunción endotelial y redujo el estrés oxidativo hasta valores normales. Este estudio sugiere que la hiperglucemia de larga duración en los pacientes con diabetes tipo 1 induce alteraciones permanentes en las células endoteliales, que pueden contribuir a producir una disfunción endotelial mediante el aumento del estrés oxidativo a pesar de que la hiperglucemia se haya normalizado40. Los resultados de este estudio se muestran en la figura 2.
 y concentraciones plasmáticas de nitrotirosina en pacientes con diabetes tipo 1 tratados con: • Insulina y and Vit. C 24h; ¿ Vit. C 24h+Insulina 12h; ¿ Insulina 24h+Vit. C 12 h Tomado de la referencia 40")
La observación de que tan sólo el control simultáneo de la glucemia y del estrés oxidativo permite normalizar la función endotelial en los pacientes con diabetes tipo 1 tiene una clara trascendencia. La evidencia existente parece sugerir la existencia de 2 vías diferentes que intervienen en la generación de la disfunción endotelial en la diabetes tipo 1: una directamente relacionada con la hiperglucemia y otra que no lo está. Una posible explicación de estas observaciones es que las 2 vías actúen de manera simultánea: una como consecuencia del nivel de glucemia que genere radicales libres durante la utilización de la glucosa en las mitocondrias, y otra por el daño permanente inducido en las células endoteliales por la hiperglucemia crónica, posiblemente a través de la glicación no enzimática de las mitocondrias.
CONSECUENCIAS TERAPÉUTICAS Y PERSPECTIVASLa evidencia que está surgiendo y que indica que la hiperglucemia deja una importante impronta en el desarrollo de futuras complicaciones tiene notables consecuencias terapéuticas: parece obligado iniciar en los pacientes diabéticos un tratamiento enérgico temprano de la hiperglucemia que presentan. Sin embargo, aunque esta estrategia puede ser más fácilmente aceptada en los pacientes con diabetes tipo 1, puede plantearse una cierta preocupación en los pacientes de tipo 2, porque este enfoque terapéutico puede incluir un uso temprano de insulina. Además, un control estricto de la hiperglucemia puede tener que incluir también el tratamiento de la hiperglucemia "posprandial"41,42, no sólo porque la hiperglucemia posprandial contribuya de manera importante a producir la HbAlc, tanto en la diabetes tipo 1 como en la tipo 243,44, sino también porque la hiperglucemia posprandial se acompaña de la formación específica tanto de especies moleculares reactivas45 como de AGE no sólo en el plasma46, sino también intracelularmente47.
Otra posible estrategia consiste en reducir la formación de AGE y la generación de estrés oxidativo concomitante con normalizaciones de la glucosa. Con varios compuestos se ha demostrado ya la capacidad de bloquear la formación de AGE. In vitro, se ha comprobado que metformina y pioglitazona previenen la formación de AGE48. Los inhibidores de la ECA y los antagonistas de receptores AT-1 son compuestos que se emplean en el control de la presión arterial; sin embargo, también son capaces de reducir la formación de AGE49. Es interesante señalar que estos fármacos actúan también como antioxidantes50 y, al menos por lo que se refiere a los antagonistas de AT-1, hay datos que indican la presencia de una acción específica frente al estrés oxidativo inducido por la hiperglucemia51. Por último, las estatinas podrían aportar también un efecto beneficioso para reducir las especies moleculares reactivas51. Combinando todos estos datos, cabría prever una futura estrategia basada en el uso de compuestos activos sobe la formación de AGE52, junto con otro compuesto capaz de actuar específicamente sobre la generación de especies moleculares reactivas mitocondriales53.
CONCLUSIONESLos nuevos datos que están apareciendo sugieren de manera uniforme que la hiperglucemia puede dejar una importante impronta en las células de los vasos sanguíneos y los órganos diana, que favorezca la futura aparición de complicaciones. Además, la evidencia existente sugiere que esta "memoria" puede aparecer a pesar de que se alcance un buen control de la glucemia. A este fenómeno se le ha denominado "memoria metabólica"6. Sin embargo, la memoria metabólica parece ser un fenómeno más común y no solamente relacionado con la hiperglucemia. Considerados conjuntamente, estos datos plantean muchas preguntas relativas al manejo terapéutico de la diabetes. Concretamente, dado que se ha demostrado ya que una intervención multifactorial enérgica reduce el riesgo de complicaciones tanto microangiopáticas como macroangiopáticas de la diabetes54, la existencia de la memoria metabólica sugiere que parece ser obligado un tratamiento enérgico muy temprano de los diversos factores de riesgo.