Keywords
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is fundamentally caused by sarcomeric gene mutations.1,2 There is great interest in genetic studies in patients with severe manifestations of the disease. These cases may be caused by high risk mutations, but they may also be caused by complex genotypes with more than one or homozygous mutations. Existing information about patients with double mutations is scarce.3-8 We present a family whose diagnosis of a homozygous mutation allowed us to improve the phenotypic interpretation of affected individuals and therefore adjust genetic counselling, respectively.
METHODS
The proband of this family belongs to a series of 130 patients with HCM monitored in a dedicated cardiomyopathy clinic, where said patients underwent gene mutation screening for the MYH7 and MYBPC3 genes. DNA was extracted from anticoagulated blood with EDTA (NUCLEON kit, Amersham Biosciences). Mutational screening was corried out by the "single stranded conformation polymorphism" (SSCP) method and fragments with abnormal mobility were sequenced. The presence of additional mutations was excluded using a genotyping platform that includes 690 previously described mutations in 15 genes (MYH7, MYBPC3, TNNT2, TNNI3, TNNC1, TPM1, ACTC, MYL2, MYL3, TTN, MYH6, TCAP, MYO6, PRKAG2, and MYLK2). All of the participants gave their informed consent to undergo genetic analysis. ´
RESULTS
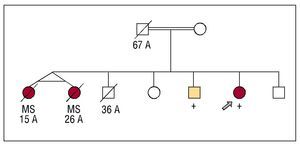
Figure 1 shows the pedigree. Two monozygous twin sisters died suddenly when they were 15 and 26 years old without having been examined. One brother died in an accident when he was 36 years old. Two affected siblings were clinically and genetically studied.

Figure 1. Family tree. Squares: males; circles: females; empty symbols: individuals without known cardiopathy; filled symbols: individuals affected by HCM or sudden death. The + symbol indicates homozygote carriers of the mutation; crossed-out marks indicate individuals that have died, with the age of death. The double line indicates co-sanguinity. The arrow shows the proband.
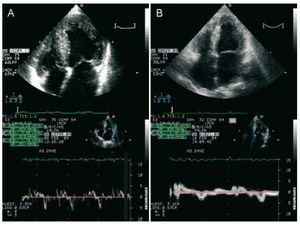
The proband is a woman who sought healthcare for an atypical precordial pain when she was 34 years old. The physical exam was normal and she presented diffuse repolarisation changes with mild negative lateral T waves in the EKG. The echocardiogram confirmed the hypertrophy of the septum, anterior and lateral walls at the basal, mid and apical levels, with a maximum thickness of 24 mm in the anterior basal septum, without sub-aortic obstruction. Her left ventricular ejection fraction (LVEF) was 50% without dilation (48 mm), severe diastolic dysfunction and left atrial enlargement (58 mm) (Figure 2). She achieved 9 MET on ergometry, with a fall in systolic arterial pressure of 30 mm Hg (from 120/70 to 90/60 mm Hg) during exercise. The patient presented periods of non-sustained ventricular tachycardia of up to 27 beats on Holter. A defibrillator was implanted due to the presence of 3 risk factors: family history of premature sudden death, non-sustained ventricular tachycardia and abnormal arterial blood pressure response during exercise. In the 6 month period after the implant, the patient had 4 episodes of prolonged non-sustained ventricular tachycardia, which were treated by the device. After increasing beta-blockers and reprogramming the defibrillator, no other treatment was needed in 4 years of follow-up. The echocardiogram when the patient was 39 years old showed a decrease in wall thickness (maximum 22 mm) and left ventricular dilatation (55 mm) with a similar ejection fraction.

Figure 2. Doppler Echocardiogram of the proband (A) and her affected brother (B). Above: apical 4 chamber view showing with left ventricular hypertrophy and dilation of both atria. Below: images of the tissue Doppler with low velocities.
The patient's brother was diagnosed when he was 28 years old in another institution during a preoperative exam. He first came to our clinic when he was 34 years old with dyspnoea of a functional class II of the NYHA, with atrial fibrillation and an incomplete right bundle branch block on EKG. He presented concentric hypertrophy without subaortic obstruction (maximum thickness, 26 mm), severe systolic and diastolic dysfunction (LVEF, 36%), with left atrial dilation (59 mm) (Figure 2). He had received amiodarone for non-sustained ventricular tachycardia that was suspended because of hyperthyroidism. A defibrillator implant was recommended due to risk factors (before the genetic diagnosis).
There was a history of consanguinity between his parents (Figure 1). His mother was examined in another centre and she was considered to have a normal phenotype (she has not come for review in our centre). His father died in an accident when he was 67 years old and he had 6 brothers, none with cardiomyopathy.
In the proband and in her affected brother, a homozygous mutation IVS6+5G>A (g5261G>A) was detected in the MYBPC3 gene, that was not previously described, that would produce a truncation of the C protein at the C2 motive level.
DISCUSSION
Approximately 5% of the cases with familial HCM present complex genetic states with more than one causal mutation: homozygotes (both alleles of the mutated gene), double heterozygotes (more than one mutation in different genes), or compound heterozygotes (more than one mutation in the same gene).3-8 In our series, the prevalence was 1.5% (the proband previously described and a compound heterozygote in MYH79). To our knowledge, only 4 homozygous mutations and 17 described carriers exist for the MYBPC3 gene.3,10-13 All have more severe phenotypes and a worse prognosis compared to simple heterozygotes. Our study is the first one to describe 2 homozygote siblings where a severe phenotype is reproduced.
The family exemplifies the effect of the gene dose in the severity of the expression of the disease. Two homozygote carriers present serious manifestations of HCM: symptoms at a young age, marked hypertrophy, systolic and diastolic dysfunction. Premature sudden death in the sisters indicates that they too may have been homozygote carriers. Given the history of consanguinity, it is important to suspect complex genetic patterns such as this one. We do not have the genotype of the parents, but they should be considered as obligatory heterozygote carriers of the mutation, probably inherited from a common ancestor. They both reached adulthood without severe manifestations of cardiomyopathy, which indicates that a single mutated allele may not be a sufficient cause to develop the disease or generate a mild phenotype with late expression.
The IVS6+5G>A mutation would affect the DNA splicing, generating aberrant transcriptions. Functional studies in mutations with similar effects demonstrated that the truncated peptide produced by the mutated allele is degraded before being incorporated into the sarcomere (haploinsufficiency). The loss of this mechanism would explain the severe expression of homozygous mutations.14 The cellular response and viability may vary depending on the quantity of mutated peptide present. Consequently, heterozygous mutations would produce hypertrophic cardiomyopathy by negative dominance over the sarcomeric function. So much that its presence in homozygosis would produce a quantity of mutated peptides that would surpass the critical threshold to cause cellular death and the loss of myocytes, leading to systolic dysfunction and ventricular dilation.15 This mechanism could be associated with the ventricular dilation and the parietal thinning observed in the follow-up of our proband and with the severe systolic dysfunction observed in her brother.
To conclude, this family illustrates the utility of genetic diagnosis in the prognostic evaluation of HCM and highlights the importance of a complete clinical evaluation of patients with this disease. Family history and the severity of the phenotype indicated the possible existence of a malignant mutation and a high risk for the carrier's descendents. The findings of a homozygous mutation in those affected radically changes our vision of prognosis in other family members. These aspects become very important when offering genetic counselling. It is expected that the descendants inherit only one copy of the mutation and thus will not develop a severe phenotype like their parents. We were not able to directly evaluate the entire family and therefore we could not adequately characterise the phenotype of the heterozygotes.
This study has been financed with a research grant from the Carlos III Health Institute Foundation of Madrid (PI 050377). Xusto Fernandez received financing from the Network of Cardiovascular Research RECAVA of the Carlos III Health Institute of Madrid. Martin F. Ortiz received financing from the BBVA-Carolina Foundation.
Correspondence:
Dr. L. Monserrat Iglesias.
Servicio de Cardiología. CHU Juan Canalejo. As Xubias, 84. 15006 A Coruña. España.
E-mail: lorenzo_monserrat@canalejo.org
Received May 13, 2008.
Accepted for publication July 2, 2008.