
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant disorder and is the most common heritable cardiomyopathy.1 Although several mutations have been related to HCM with poor prognosis, the prognostic value of specific mutations in HCM remains controversial. Genetic variants in the β-myosin heavy chain encoded by MYH7 involving the so-called “converter region” have been associated with early disease and higher rates of transplant and malignant arrhythmias.2 The converter domain extends from Phe-709 to Arg-777 and is believed to be a key element of the actomyosin complex where elastic distortion occurs.
We describe a family with a previously undescribed genetic variant in the converter region in MYH7 (c.2212A> C; p.Ser738Arg). The index case is a 42-year-old woman diagnosed with HCM when she was 24 years old and who was admitted to the cardiology ward due to heart failure. An echocardiogram showed severe biventricular systolic dysfunction. Although she received specific treatment for heart failure (table 1), at the age of 45 years she required a heart transplant.
Patients’ clinical characteristics
| Patient | Birth year | Age at HCM diagnosis, y | Maximum myocardial thickness | LVOTgradient | Mitral regurgitation | LVEF | LGE | Symptoms | Arrhythmia | Treatment | Implantable devices | Final outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(index case) | 1960 | 24 y(1984) | > 18 mm | NA | Moderate (functional) | <30% | NA | Syncope(29 y)Heart failure(42 y) | Atrial fibrillation | ACEISotalolDigitalisWarfarinLoop diuretics | ICD – CRT(2002) | Transplant(45 y) |
| 2 | 1990 | 22 y(2012) | 18 mm | 48 mmHg | Moderate(SAM) | 61% | Yes | None | NSVT | Atenolol | ICD(2015) | Follow-up |
ACEI, angiotensin-converting enzyme inhibitor; CRT, cardiac resynchronization therapy; HCM, hypertrophic cardiomyopathy; ICD, implantable cardiac defibrillator; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract; NA, not available; NSVT, nonsustained ventricular tachycardia; SAM, systolic anterior motion.
A genetic test using new generation sequencing of a cardiomyopathies general panel including 17 genes (ACTC, DES, FLNC, GLA, LAMP2, MYBPC3, MYH7, MYL2, MYL3, PLN, PRKAG2, PTPN11, TNNC1, TNNI3, TNNT2, TPM1, and TTR) was performed using an Illumina Hiseq sequencer. Alignment and filtering of variants was performed in a custom in-house pipeline. Variant pathogenicity was classified according to the American College of Medical Genetics and Genomics recommendations.
A missense genetic variant in MYH7 (c.2212A> C; p.Ser738Arg) was identified. This genetic variant had neither been previously described nor reported in general population databases. The variant affects a conserved amino-acid in the “converter region” of the protein. Three other variants have been described in the same residue in association with HCM (p.Ser738Asn, p.Ser738Arg and p.Ser738Thr). In silico predictors (SIFT, Polyphen-2) consider this variant probably deleterious. The variant was considered likely pathogenic.
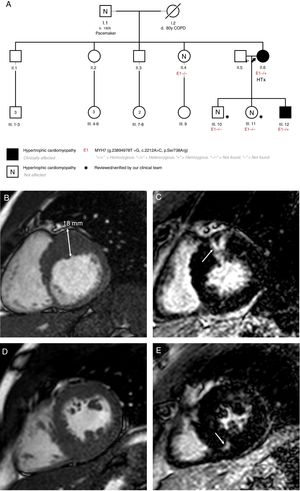
Family screening showed that the patient had 2 (a male and a female) healthy children with negative genetic testing. Her third child, a male diagnosed with HCM when he was 22 years old, carried the same mutation. The patient's mother died before HCM diagnosis. Her father is currently alive and HCM has been ruled out with echocardiogram, although he declined to provide consent for genetic testing. In one of the patient's sisters, HCM was ruled out after complete evaluation and genetic testing. The other three siblings refused evaluation. The family pedigree is shown in figure 1A.
. D: case 2, cardiac magnetic resonance. Short axis-papillary muscles. E: case 2, cardiac magnetic resonance. Late gadolinium enhancement in the mid inferoseptal segment (arrow). COPD, chronic obstructive pulmonary disease; HTx, heart transplant.")
A: patients’ familial pedigree. Black arrow points to index case. B: case 2, cardiac magnetic resonance. Short axis-basal. C: case 2, cardiac magnetic resonance. Late gadolinium enhancement in the basal anterior segment (arrow). D: case 2, cardiac magnetic resonance. Short axis-papillary muscles. E: case 2, cardiac magnetic resonance. Late gadolinium enhancement in the mid inferoseptal segment (arrow). COPD, chronic obstructive pulmonary disease; HTx, heart transplant.
The son of the index case was diagnosed with HCM when he was 22 years old. He is currently 28 years old. Severe hypertrophy has been documented (maximal wall thickness of 18mm) with normal ventricular function and basal left ventricular outflow tract obstruction (48mmHg despite atenolol 100mg twice a day) and moderate mitral regurgitation secondary to systolic anterior motion of the mitral valve. Cardiac magnetic resonance showed intramyocardic late gadolinium enhancement (LGE) in the basal anterior segment and in the mid inferoseptal segment (figure 1B-E). Holter-electrocardiogram showed nonsustained ventricular tachycardia. Estimated sudden cardiac risk was 6.65%1 and an ICD was implanted when he was 25 years old. Sanger sequencing showed that this patient was also a carrier of p.Ser738Arg.
Guidelines for HCM recommend long-term follow-up for healthy carriers of genetic variants.1 However, recommendations based on specific variants are still lacking. Identification of variants related to worse prognosis is key to taking personalized therapeutic decisions.
García-Gustiniani et al.3 reported a cohort of 117 patients with mutations in the converter region and described low and high-risk variants.
We describe a variant in the MYH7 converter region related to HCM with early development of myocardial hypertrophy and fibrosis, and rapid progression to systolic dysfunction and heart failure.
Almost 3% of HCM patients develop systolic dysfunction. Early identification of these high-risk patients is mandatory. Family history of end-stage HCM, diagnosis at a young age, ventricular tachycardia and the presence and the extension of LGE can predict the development of systolic dysfunction in HCM.4 Mutations in the MYH7 converter region have been associated with early development of HCM and an increased incidence of left ventricular dysfunction.3 The same region has also been associated with a phenotypic spectrum including dilated, noncompacted and restrictive cardiomyopathies with some individuals showing overlapping features.3
Our index case was diagnosed with HCM at a young age and showed rapidly progressive systolic dysfunction requiring heart transplant. Her son was diagnosed at the age of 22 years, had an elevated high-risk for SCD and received an implantable cardiac defibrillator when he was 25 years old. In addition, he has several risk factors for early systolic dysfunction.
In conclusion, genetic testing seems to be an adequate tool to assess prognosis in HCM, although it is still underused. The c.2212A> C; p.Ser738Arg mutation in the MYH7 converter region may be a marker of rapid progression to end-stage HCM and may also confer an increased risk of SCD. Future studies and reports would help to increase our knowledge of this field and to offer personalized follow-up.
.