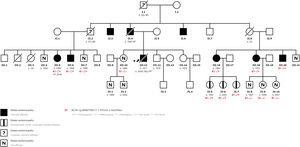

Dilated cardiomyopathy (DCM) is a myocardial disease typified by left ventricular or biventricular dilation and dysfunction that cannot be explained by abnormal loading conditions, and which provokes heart failure (HF), arrhythmias, or sudden cardiac death (SCD). A familial association has been demonstrated in a high percentage of cases, highlighting the importance of genetic studies of family members. We present a family with a previously undescribed mutation in the ACTA1 gene. Informed consent were obtained from all patients and approval was obtained from the ethics committee. Figure 1 shows the family tree.

The index case is a 36-year-old man, with bronchial asthma as the only relevant medical history. His family history includes a paternal grandfather and uncle with SCD, and his father and 2 other paternal uncles with DCM. Onset was a first episode of HF in April 2005. During admission, the patient was diagnosed with DCM, with an echocardiogram showing left ventricular dilation with severely depressed global systolic function and severe mitral regurgitation. Coronary disease was ruled out and, despite treatment optimization, symptoms persisted. The patient underwent follow-up at our HF and Inherited Heart Disease Unit, with progressive deterioration of left ventricular ejection fraction (LVEF) and, in July 2008, he died after rapidly progressing cardiogenic shock.
In 2019, a 55-year-old man, a cousin of the index patient, was referred to our unit (figure 1, case III.7) for familial screening. His sister (figure 1, case III.5), who was under follow-up at another center, experienced onset of DCM in 2008, and had an implantable cardioverter-defibrillator that produced appropriate shocks during follow-up. A massive parallel sequencing of 121 genes found no presence of pathogenic mutation but did uncover a variant of uncertain significance in the ACTA1 gene. This patient rapidly developed advanced HF, requiring a heart transplant in the same year as diagnosis.
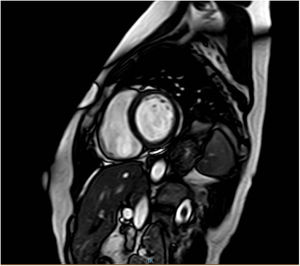
During the same period, a 47-year-old woman attended our clinic (figure 1, case III.16). The patient was the cousin of the index case and cases III.5 and III.7. Cardiac diagnosis tests found a mildly depressed left ventricular ejection fraction (46%) without ventricular dilation, and magnetic resonance imaging showed the presence of linear midmyocardial late gadolinium enhancement of the anteroseptal-septal-inferoseptal and medial basal segments (figure 2).

Cardiac diagnosis tests of case III.7 found no evidence of structural heart disease and targeted genetic testing of both this patient and case III.16 found both to be carriers of the heterozygous variant NM_001100.3:c.757G>A; p. (Gly253Ser) of the ACTA1 gene, which coincided with the mutation previously described in the transplanted patient. During the following year, the familial screening was extended to include first-degree relatives, among whom the most outstanding finding was the 3 daughters of case III.16, who are all ACTA1 mutation carriers but do not currently have associated structural heart disease.
The ACTA1 gene encodes the alpha-actin isoform, a sarcomeric protein for muscular contraction particularly abundant in skeletal muscles. Mutations in ACTA1 have classically been associated with different types of skeletal myopathy. Although cardiac involvement is infrequent, some published cases associate ACTA1 with cardiac involvement in the form of DCM1 and hypertrophy2,3 occurring jointly with myopathy. It is striking that the predominant presentation is hypertrophic cardiomyopathy in the pediatric age and DCM in adults.
Nevertheless, we find very few records of mutations in this gene with myocardial involvement alone. The study by Carnevale A et al.,4 conducted with 55 DCM patients who underwent broad genetic testing, found that only 1 of the patients was a carrier of an ACTA1 gene mutation, although myopathic involvement was not stated. To date, there is a single publication that describes a pathogenic mutation of ACTA1 with myocardial involvement alone,5 specifically the heterozygous mutation p.Arg256His, which is expressed as DCM, ventricular arrhythmia, and SCD. Our work describes a new mutation in ACTA1, NM_001100.3:c.757G>A; p. (Gly253Ser), possibly associated with DCM without concomitant skeletal myopathy. This is a missense-type variant that affects an amino acid located at the domain level of the protein that is relevant to actin folding. Furthermore, the variant described by Reza et al.5 (p.Arg256His) is located very close to this new variant (p.Gly253Ser)—only 3 amino acids apart in the protein—giving rise to the hypothesis that variants in this region could be related to this phenotype, thus opening the door to new research. To date, the literature has not described this variant in association with the development of disease, nor does it appear in the databases we used as control (ACTA1-LOVD variants database, ClinVar, Exome Variant Server, Exome Aggregation Consortium and the Genome Aggregation Database).
The pathogenicity of this variant was initially uncertain but this study of a large part of the family shows there is cosegregation of this variant with the phenotype. This work may lead to further study, bearing in mind the penetrance, unfavorable progression, and dire prognosis among most described cases, for whom early diagnosis and treatment are of particular interest. We also wish to highlight the importance of conducting genetic testing and familial studies to verify cosegregation. Despite the development of these tools in recent years, they were not easily affordable at the time of onset in our family index case; had they been more generally available, a more precise diagnosis would have had favorable repercussions for the other family members. Finally, as other research has also highlighted, there is a need to broaden knowledge in this field because it is striking that, despite alpha-actin being one of the most expressed proteins in skeletal muscles, it can produce myocardial involvement alone.
FUNDINGNo funding was received for the study.
AUTHORS’ CONTRIBUTIONSA. Díaz Expósito: collection of patients, complementary tests and genetic study; literature review; image search; article drafting. A. Robles Mézcua: review of genetic studies; development of family tree; article drafting. I. Pérez Cabeza and J. M. García Pinilla: scientific letter review.
CONFLICTS OF INTERESTNo conflicts of interest.