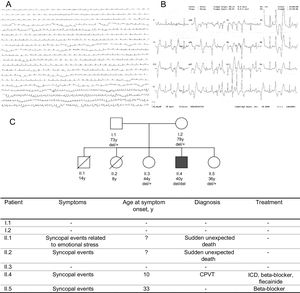
We present the case of a 40-year-old man with a history of syncope since childhood. He had been treated with carteolol 2.5mg bid from the age of 10 years due to syncope and ventricular arrhythmia. When he was aged 17 years, the beta-blocker was changed and nadolol was started at 80mg qd, which the patient voluntarily discontinued. He was admitted to hospital due to syncope while he was walking. A strong family history of sudden cardiac death was observed: a brother and a sister suddenly died at the age of 8 and 14 years, respectively. The parents were apparently healthy and the grandparents died in old age from tumors. The results of laboratory tests, electrocardiography, QT interval, echocardiographic examination, and cardiac magnetic resonance imaging were normal. A nonsustained polymorphic ventricular tachycardia (PVT) was detected in electrocardiogram recordings. Treadmill testing revealed nonsustained PVT during peak effort that disappeared during rest (Figure 1A and Figure 1B). A diagnosis of catecolaminergic polymorphic ventricular tachycardia (CPVT) was made due to an exercise-induced PVT in the presence of a structurally normal heart and normal electrocardiogram. Beta-blocker treatment was restarted and a treadmill test was repeated. An implantable cardioverter-defibrillator with long delays before shock delivery was implanted due to persistence of nonsustained PVT during exercise. During follow-up, flecainide was added at a dose of 100mg bid to beta-blocker treatment due to appropriate shocks. After 6 months follow-up, ventricular arrhythmia control was notably improved with no sustained episodes.
. B: 12-lead ECG during treadmill testing with runs of PVT. C: family pedigree. The genotype for the CASQ2 deletion is indicated. CPVT, catecholaminergic polymorphic ventricular tachycardia; ECG, electrocardiogram; ICD, implantable cardioverter-defibrillator.")
A: ECG telemetry recording showing highly frequent ventricular ectopy (bottom). B: 12-lead ECG during treadmill testing with runs of PVT. C: family pedigree. The genotype for the CASQ2 deletion is indicated. CPVT, catecholaminergic polymorphic ventricular tachycardia; ECG, electrocardiogram; ICD, implantable cardioverter-defibrillator.
A genetic test was performed to confirm the diagnosis.1–3 We collected a peripheral blood sample from the index case and 4 relatives (Figure 1C). Unfortunately, biological samples from deceased relatives were not available. Massive sequencing for 28 genes associated with arrhythmogenic disease was performed in the index case.4
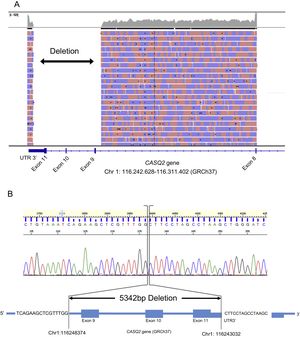
Sequencing showed no causal single nucleotide variant but did show the absence of the sequence for the last 3 exons of the CASQ2 gene. Long-range polymerase chain reaction was conducted to amplify a fragment of approximately 18kb, where the deletion is located. The results confirmed the presence of a fragment of approximately 13kb in the index case and a fragment of approximately 18kb in a control sample. Long-range polymerase chain reaction product of the index case was sequenced with the Ion Proton System (Thermo Fisher Scientific). The results demonstrated the presence of an approximately 5kb deletion causing the loss of exons 9, 10, and 11 of the CASQ2 gene (Figure 2). The exact location of the deletion was confirmed by using specific polymerase chain reaction and Sanger sequencing (NM_001232:c.839-461_*830del).
 visualization for long-range PCR product of the index case. B: electropherogram and representation of the exact location of the deletion.")
Our family index case showed a homozygote deletion classified as pathogenic according to the American College of Medical Genetics (ACMG),5 confirming the CPVT diagnosis, whereas unaffected family members were heterozygous carriers of the deletion (Figure 1C). One of the sisters had recently experienced syncope of unknown cause. Based on the hypothesis of a higher risk of arrhythmias associated with heterozygous variants, she was treated with beta-blockers.
Catecolaminergic PVT is an inheritable arrhythmogenic disorder characterized by adrenergic-induced bidirectional ventricular tachycardia or PVT. Two genetic types have been identified: a dominant variant due to mutations in the cardiac ryanodine receptor gene (RyR2) and a rare recessive variant caused by mutations in the cardiac calsequestrin gene (CASQ2).1,3 Mutations in other genes such as KCNJ2, ANK2, TRDN, and CALM1 have been identified in patients with clinical features similar to CPVT but it is not clear whether they are phenocopies of CPVT.3 Most of the CASQ2 gene mutations described are truncating or splicing genetic variants and to date there have been no reports of causal copy number variations. Although some copy number variations have been described in channelopathies, they are not common; in the specific case of CPVT, a deletion of exon 3 of RyR2 has been described.
The clinical manifestations occur in the first decade of life and are triggered by physical activity or emotional stress. First-line therapy consists of exercise restriction and beta-blockers without sympathomimetic activity. Preliminary data suggest that flecainide reduces the ventricular arrhythmia burden in some patients and should be considered as an addition to beta-blockers.3 Diagnosis is confirmed by genetic testing. Mass sequencing technologies allow rapid screening of single nucleotide or small indel variants. However, the presence of large deletions or insertions may go unnoticed, if we only take into account the lists of variants reported by variant calling programs. The presence of these insertions or deletions must be checked with specific programs, otherwise the sequencing results should be reviewed with genomic visors, checking the coverage of all target regions to be sequenced.4
This is the first reported case of a copy number variation as a cause of CVPT in a nonconsanguineous family with poor prognosis, with a severely affected index case carrying the variant in homozygosis and with 2 siblings who died suddenly at very early ages.
FundingThis work was supported by Plan Estatal de I+D+i 2008-2011 and 2013-2016, Subdirección General de Evaluación y Fomento de la Investigación (ISCIII-SGEFI) from Instituto de Salud Carlos III (ISCIII) and Fondo Europeo de Desarrollo Regional (FEDER) (grant numbers PI16/00903, RD12/0042/0037, CB16/11/00226).