Arrhythmogenic right ventricular dysplasia (ARVD) is a genetic disease with a mutation rate of 50% to 60% in desmosomal genes. The disease is usually inherited in an autosomal dominant pattern, except for Naxos disease and Carvajal syndrome, which have specific phenotypic characteristics (palmoplantar keratoderma, wooly hair, etc) and are inherited in an autosomal recessive pattern. Since the first report of an association between the desmoglein-2 gene (DSG2) and the development of ARVD in 2006,1 numerous mutations have been described, almost all of which are heterozygous with autosomal dominant behavior. This gene can be inherited as compound or digenic heterozygosis.2 It has recently been shown that mutations in this gene can exhibit an autosomal recessive pattern in the absence of cardiocutaneous syndrome, although the available data on this issue are very scarce.3,4 We describe the clinical spectrum and autosomal recessive mode of transmission of a double variant in DSG2 in the absence of cardiocutaneous syndrome.
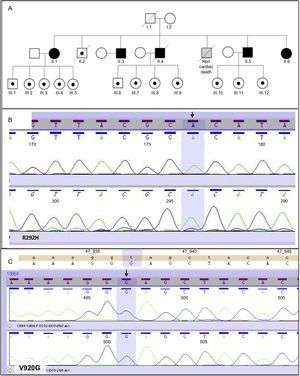
This study included 18 participants from the same family. Figure A shows the family tree. Five participants were homozygous for mutations R292H and V920G in DSG2 and all 5 were second-generation family members. The remaining 13 participants were heterozygous for both mutations. Of these participants, 12 were third-generation family members and 1 (participant II.2) was from the second generation. The R292H mutation (Figure B) in DSG2 is responsible for an amino acid change in which arginine is substituted by histidine in the third extracellular domain of the amino (N)-terminal region. The V920G variant is responsible for an amino acid change that replaces valine with glycine (Figure C) in the C-terminal cytoplasmic tail.3
 and heterozygous participants (white with a black dot). Squares and circles symbolize men and women, respectively. B: Homozygous variant R292H on DSG2 (arrow). C: Homozygous variant V920G on DSG2 (arrow).")
All participants provided clinical information on events such as syncope, ventricular arrhythmias, automatic cardioverter-defibrillator discharges, hospitalization for heart failure, and functional class. All participants underwent 12-lead electrocardiography, echocardiography, and, when possible, cardiac magnetic resonance imaging, and 24-hour Holter monitoring.
Statistical comparisons were performed between homozygous and heterozygous participants with both variants. Differences in clinical events were analyzed using the Fisher exact test (SPSS 19.0; Chicago, United States).
The mean age of the homozygous participants was higher than that of the heterozygous participants, because all the homozygous participants were second-generation family members, whereas only 1 of the 13 heterozygous participants was second-generation (participant II.2; Figure A). No differences were found by sex.
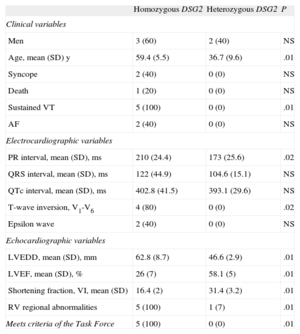
All the homozygous participants had a clinical picture compatible with arrhythmogenic dysplasia with biventricular involvement, severe ventricular arrhythmias, and severely impaired left ventricular function. A statistically significant difference was found between groups because none of the heterozygous participants met the diagnostic criteria for ARVD. None of the 5 homozygous participants had the phenotypic features characteristic of Naxos disease or Carvajal syndrome, and all 5 needed an automatic implantable cardioverter-defibrillator. The statistical comparisons of the clinical, electrocardiographic, and echocardiographic data by group are shown in the Table.
Statistical Comparison of Clinical Data Between Homozygous and Heterozygous Participants
| Homozygous DSG2 | Heterozygous DSG2 | P | |
| Clinical variables | |||
| Men | 3 (60) | 2 (40) | NS |
| Age, mean (SD) y | 59.4 (5.5) | 36.7 (9.6) | .01 |
| Syncope | 2 (40) | 0 (0) | NS |
| Death | 1 (20) | 0 (0) | NS |
| Sustained VT | 5 (100) | 0 (0) | .01 |
| AF | 2 (40) | 0 (0) | NS |
| Electrocardiographic variables | |||
| PR interval, mean (SD), ms | 210 (24.4) | 173 (25.6) | .02 |
| QRS interval, mean (SD), ms | 122 (44.9) | 104.6 (15.1) | NS |
| QTc interval, mean (SD), ms | 402.8 (41.5) | 393.1 (29.6) | NS |
| T-wave inversion, V1-V6 | 4 (80) | 0 (0) | .02 |
| Epsilon wave | 2 (40) | 0 (0) | NS |
| Echocardiographic variables | |||
| LVEDD, mean (SD), mm | 62.8 (8.7) | 46.6 (2.9) | .01 |
| LVEF, mean (SD), % | 26 (7) | 58.1 (5) | .01 |
| Shortening fraction, VI, mean (SD) | 16.4 (2) | 31.4 (3.2) | .01 |
| RV regional abnormalities | 5 (100) | 1 (7) | .01 |
| Meets criteria of the Task Force | 5 (100) | 0 (0) | .01 |
AF, atrial fibrillation; DSG2, desmoglein-2 gene; LV, left ventricle; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; RV, right ventricle; Task Force, Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology; VT, ventricular tachycardia.
Unless otherwise indicated, the data are expressed as No. (%).
The 13 participants who were heterozygous for both mutations had a normal electrocardiogram and no clinical events. One second-generation heterozygous participant (II.2 participant; Figure A) had ischemic heart disease, but had no signs of arrhythmogenic dysplasia.
This study is the first to describe a double homozygous variant with autosomal recessive behavior in a desmosomal gene, DSG2, as a cause of ARVD with severe clinical expression. The absence of findings characteristic of Carvajal syndrome and Naxos disease provides supporting evidence that ARVD may manifest as an autosomal recessive disease with a complex genotype in the absence of associated cardiocutaneous syndromes. These results indicate the need for caution in clinical practice when interpreting heterozygous variants in patients without phenotypes characteristic of the disease.5
Of the 2 homozygous variants described in this study, the one shown in Figure B has not been described, has rarely been observed in the general healthy population, and has a high probability of altering the protein architecture of the desmosome. The other variant, V920G, affects an interspecies conserved residue and has been found to be absent in more than 200 healthy controls. It has been previously described as a potential cause of dilated and arrhythmogenic cardiomyopathy, although it is usually found in association with other heterozygous variants in DSG2 or in another desmosomal gene such as the DSP gene. The pathogenic potential of V920G alone remains unclear.
In conclusion, this study describes an autosomal recessive mode of transmission with 2 homozygous variants on DSG2 as a cause of ARVD with severe biventricular involvement in the absence of clinical signs of cardiocutaneous syndrome. It cannot be definitively affirmed that the interaction of the 2 mutations is the cause of the severity of this recessive phenotype, but it may be largely due to the pathogenic effect of R292H modulated by V920G.