Molecular characterization of congenital heart diseases now includes the not infrequent dysmorphic Noonan syndrome. A study of 6 genes of the RAS-MAPK pathway in Spanish patients is presented: the impact of heart disease, clinical expressivity, and diagnostic yield are investigated.
MethodsThe study included 643 patients (and 182 family members) diagnosed by dysmorphologists, cardiologists, and pediatric endocrinologists from 74 tertiary hospitals. Bidirectional sequencing analysis of PTPN11, SOS1, RAF1, BRAF, KRAS and HRAS focused on exons carrying recurrent mutations accounting for 80% to 95% of previously described mutations.
ResultsMutations were detected in 230 patients (91 women and 139 men) in 200 (31%) families (172 PTPN11+, 14 SOS1+, 9 RAF1+, 5 BRAF+). There was specific reference to the heart defect suffered in 156 index cases: 103 patients had shown pulmonary stenosis, 12 pulmonary stenosis with hyperthrophic cardiomyopathy, 18 hypertrophic cardiomiopathy, and 14 other cardiopathies; heart disease was absent in 9 index cases. Heart disease had not been documented in 23 of 30 family members with positive genotype and compatible clinical signs. Diagnostic yield was higher (P=.016) for samples from some centers (53%; 14/32) and even from certain professionals (64%; 9/14; P=.019). Characterization rate was 18% in patients for whom clinical data were not available. Genotyping led to a more precise diagnosis in 26 patients.
ConclusionsMost patients (94%) with a positive genotype had known congenital heart disease, 79% pulmonary stenosis and 12% hyperthrophic cardiomyopathy. Cardiopathy had not been documented in 76% of family members carrying the mutation. Molecular study is a useful tool in these syndromes but a more rigorous clinical diagnosis should be intended as well.
Keywords
.
IntroductionNoonan syndrome1 (NS) (OMIM 163950) is considered a relatively common hereditary syndrome in the population (1:2500). Its clinical signs include a characteristic facial dysmorphism, short stature, thick neck and chest deformity, cryptorchidism in males, and heart disease2, 3, 4 (usually pulmonary valve stenosis [PS] as well as hypertrophic cardiomyopathy [HCM]), and Van der Burgt's criteria5 are generally accepted for diagnosis. It is a dominant monogenic entity which often appears de novo.
NS has gone from being interpreted as the “male Turner” to being recognized as a syndrome stemming from impaired monoallelic point (dominant) autosomal genes. The PTPN11 gene mutations in 12q24.1, which encodes a phosphatase SHP2, are the first changes reported6 in approximately 50% of patients. Some PTPN11-negative patients (2%-5%) have KRAS mutations (12p12.1),7, 8 which provided evidence that changes in the RAS-MAPK pathway could lead to the syndrome. This regulatory pathway, initially known for its involvement in cancer, controls the proliferation/apoptosis balance and cell migration and, thereby, morphogenesis. Unlike oncogenic mutations, which are of the somatic type and are limited to the affected clone, the alteration in the dysmorphic syndrome occurs in the germ line and is compatible with the individual's viability. The variants that cause one condition or another usually differ.8
Recent studies have established that alterations in other protein-encoding genes in the RAS-MAPK pathway, the RAF1 (at 3p25),9, 10 BRAF (at 7q34),11 and MEK-1 (at 15q21) kinases,12 and the SOS1 guanine nucleotide-exchange protein (in 2p22-p21)13, 14 also contribute to the molecular basis of NS. The SOS1 gene, although it was described later, has moved into second place in terms of its contribution to NS, at 20% of negative PTPN11.15 Mutations have also recently been documented in NRAS (1p13.2),16 although at a much lower frequency (0.4%).
At the same time, it became evident that several syndromes showing some phenotypic overlap with NS, such as LEOPARD17, 18, 19 (OMIM 151100), the cardio-facio-cutaneous syndrome12, 20 (CFC, OMIM 115150), Costello syndrome21 (OMIM 218040), neurofibromatosis type I22 (OMIM 162200), and Legius syndrome (OMIM 611431) were also due to alterations in this pathway, thereby extending the involvement to HRAS (at 11p15.5), SPRED (at 15q13.2), and MEK-2 (at 19p13 .3). All of these dysmorphic syndromes showed dominant and recurrent mutations, and different names have been proposed to characterize this group of entities, including “RAS-MAPK syndromes”, “neuro-cardio-facio-cutaneous syndromes2”, and “rasopathies”.23, 24 There are associations between some clinical signs and the genes involved which may help stratify the molecular analysis.25, 26, 27, 28, 29, 30, 31
The objectives of this study were to investigate the genotypic profile and the diagnostic yield of molecular analysis in Spanish patients with Noonan and related syndromes, and to assess the impact of heart disease and clinical expression in proven mutation carriers.
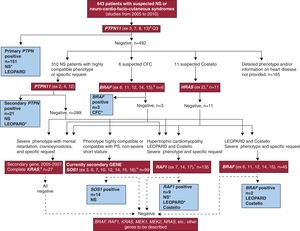
Methods PatientsThis paper reports the results of molecular analysis carried out between January 2005 and December 2010 in 643 patients (397 men and 246 women) with suspected NS or a related neuro-cardio-facio-cutaneous syndrome (Figure 1). The analyses were requested by specialists responsible for the patients (usually cardiologists, dysmorphologists, pediatric endocrinologists, or geneticists; less frequently, neurologists or neonatologists). The same specialist requesting the analyses also performed genetic counseling before and after the test and obtained informed consent. The samples came from 74 hospitals in 11 autonomous communities. The pre-test form offered to all centers (supplementary material) was based on that used by Zenker et al.27 in the study with the most rigorous phenotypic selection and highest quality of genotypic characterization.

Figure 1. Flow diagram illustrating the genetic study of the RAS-MAPK pathway performed in patients. Genes studied, clinical signs, and/or suspected syndrome in patients and number of samples submitted for each study over the 2005-2010 period. The genes analyzed are in red boxes and brackets indicate recurrent exons analyzed. The blue boxes indicate the number of carrier patients detected and the syndromes they presented, the most frequent are marked with an asterisk (*). The primary study of PTPN11 was applied in all samples; patients without a detailed phenotype and/or heart disease data for the period studied were excluded from further analysis, although they were included for the calculation of the overall diagnostic yield for molecular analysis. aThe primary study of PTPN11 (4 exons, to which 86% of the mutations described for this locus are attributed 6 ) was performed on all samples, and the secondary study (3 additional exons, covering a total of 99% of mutations described in the international literature 25–28 ) was systematically applied to patients with a phenotype and heart diseases such as pulmonary valve stenosis or hypertrophic cardiomyopathy, and patients with LEOPARD syndrome. Other genes could be analyzed if considered appropriate, as shown in the diagram: bBRAF was analyzed in patients with cardio-facio-cutaneous syndromes, and secondarily in patients with Costello syndrome, in PTPN11-negative LEOPARD patients, and in RAF1. cIn patients with Costello syndrome HRAS analysis was the initial study and was directed at Gly12 and Gly13, which are mutated in 85-90% of these patients. 21 dIn an early stage of the study, KRAS was the second gene analyzed in Noonan syndrome, Costello syndrome, and cardio-facio-cutaneous syndrome. However, it was displaced by eSOS-1 (Noonan syndrome) and fRAF1 (patients with HCM, LEOPARD syndrome, and Costello) in our secondary studies. CFC, cardio-facio-cutaneous syndrome; PS, pulmonary valve stenosis; SN, Noonan syndrome.
Subjects were mainly pediatric patients; 95% were under 20 years of age (8.58 [17.2] years). We analyzed 182 members of 98 families (15%). Mutation was found in 230 patients, 30 of whom were related. Of the 196 mutation-carrying patients, clinical data on the specific type of heart disease suffered was available for 156 (9 patients had no heart disease). In order to avoid bias in data availability, and a possible increase in subjectivity in the clinical examination of patients with mutation, no further information was requested once the molecular data had been obtained, even when we were informed of modifications in the clinical diagnosis of some patients.
For the molecular study, the 2 types of sample typically used were blood anticoagulated with ethylenediaminetetraacetic acid and DNA extracted by the requesting hospital. Other tissues (liver and kidney tissue obtained during necropsy) were very occasionally analyzed, and there were 4 prenatal samples.
Molecular Genetic StudyThe molecular study began in 2005 with the analysis of PTPN11. Genes described later were also incorporated into the study as shown in Figure 1, which details the process of genotyping. This diagram should not be considered definitive because the molecular basis of these syndromes is not fully known. Likewise, the series is not exhaustive in terms of the genes analyzed and is still open to data for the stratification and characterization of new cases.
The number of genes analyzed per patient was: 2 genes in 81 patients, 3 genes in 38 patients, 4 genes in 15 patients, 5 genes in 6 patients, and 6 genes in 1 patient. In the 182 family members, the study addressed the mutated exon in the index patient of the corresponding family, to detect or rule out alteration. Sequencing was performed bidirectionally using fluorescent dideoxynucleotides and ABI Prism® 3100 (Instrumental Line Sequencing, Hospital General Universitario Gregorio Marañón). The results were analyzed with SeqScape 2.5. Detected abnormalities not previously described were evaluated in silico using MutPred32 version 1.2 (Buck Institute, Indiana University; http://mutpred.mutdb.org) and SIFT33 version 4.0.3 (J. Craig Venter Institute; http://sift.jcvi.org). These programs explore the evolutionary conservation and functionality of the mutated protein (deduced from alterations in folding, the interaction of neutral or charged amino acid residues, and other features of the functional models) and allow us to infer the potential pathogenic nature of a change of sequence.
Data were analyzed using the Epidat 3.1 test for comparison of proportions.
Results GenotypingTable 1 shows the alterations detected in 200 Spanish patients (126 men and 74 women) and the most frequently associated types of heart disease. The series includes both the de novo patients and the index patients for detected familial forms. The sex distribution of mutation carriers reflects the bias found in the original sample. Figure 1 diagrams the applied study and the syndromes associated with the different mutations.
Table 1. Alterations in RAS-MAPK Genes Detected in 200 Index Patients With Noonan, and Other Neuro-Cardio-Facio-Cutaneous Syndromes. De Novo Cases and Familial Forms. Genotype and Heart Disease.
| Index patients (no. of cases) | ||||||||||
| Gene | Protein domain | Exon | Genotype a | Mutation carriers | Frequency distributions b | De novo form | Familial form | Heart disease c | Heart disease present | No heart disease |
| PTPN11 | Regulatory domain | 2 | p.Thr42Ala | 4 | 2% | 4 | 0 | PS* | 3 | |
| 3 | p.Thr52Ile | 1 | 1% | 0 | 1 | PS | 1 | |||
| 3 | p.Asn58Asp, p.Asn58His, Asn58Lys | 5 | 3% | 5 | 0 | PS* (3), other | 4 | |||
| 3 | p.Gly60Ala, p.Gly60Ser | 4 | 2% | 4 | 0 | PS | 2 | 1 | ||
| 3 | p.Asp61Gly, p.Asp61Asn | 8 | 5% | 8 | 0 | PS* (5), other | 7 | 1 | ||
| 3 | p.Tyr62Asp | 2 | 1% | 2 | 0 | PS* (1), other | 2 | |||
| 3 | p.Tyr63Cys d | 10 | 6% | 7 | 3 | PS* (5), other | 6 | |||
| 3 | p.Ala72Gly, p.Ala72Ser | 9 | 5% | 9 | 0 | PS* (6), HCM | 8 | |||
| 3 | p.Thr73Ile, p.Thr73Leu | 3 | 2% | 3 | 0 | PS* | 1 | 1 | ||
| 3 | p.Glu76Asp | 1 | 1% | 1 | 0 | |||||
| 3 | p.Gln79Arg | 9 | 5% | 7 | 2 | PS (5) | 5 | |||
| 3 | p.Asp106Ala | 3 | 2% | 3 | 0 | PS* | 3 | |||
| Bridge domain | 4 | p.Glu110Lys | 1 | 1% | 1 | 0 | ||||
| 4 | p.Glu139Asp | 6 | 3% | 6 | 0 | PS* | 5 | 1 | ||
| Active site domain | 7 | insCAA(Gln256) e | 1 | 1% | 0 | 1 | ||||
| 7 | p.Gln256Arg | 1 | 1% | 1 | 0 | PS | 1 | |||
| 7 | p.Leu261Phe | 1 | 1% | 1 | 0 | |||||
| 7 | p.Gly268Cys | 1 | 1% | 1 | 0 | 1 | ||||
| 7 | p.Tyr279Cys | 5 | 3% | 4 | 1 | PS (1), HCM | 2 | |||
| 7 | p.Ile282Val | 3 | 2% | 3 | 0 | PS* (1), other | 2 | |||
| 7 | p.Phe285Leu, | 5 | 3% | 4 | 1 | PS* (2), other | 3 | |||
| 8 | p.Phe285Cys, p.Phe285Ser | 3 | 2% | 3 | 0 | PS* | 2 | |||
| 8 | p.Asn308Asp, p.Asn308Ser | 56 | 33% | 53 | 3 | PS* (33), HCM, other | 37 | 3 | ||
| 12 | p.Ala461Ser, p.Ala461Thr | 2 | 1% | 2 | 0 | HCM* | 2 | |||
| 12 | p.Thr468Met | 9 | 5% | 8 | 1 | PS* (5), HCM* | 9 | |||
| 13 | p.Pro491Ser, p.Pro491Thr | 4 | 2% | 3 | 1 | PS* | 3 | |||
| 13 | p.Ser502Ala, p.Ser502Leu | 2 | 1% | 2 | 0 | PS* | 2 | |||
| 13 | p.Gly503Arg | 1 | 1% | 1 | 0 | PS* | 1 | |||
| 13 | p.Met504Val | 10 | 6% | 8 | 2 | PS* (5), HCM | 7 | |||
| 13 | p.Gln510Glu, p.Gln510Pro, p.Gln510Arg | 3 | 2% | 2 | 1 | PS (1), HCM | 2 | 1 | ||
| SOS1 | DH domain | 6 | p.Thr266Lys | 2 | 11 | 3 | PS* | 2 | ||
| 6 | p.Met269Thr | 2 | PS* | 2 | ||||||
| 7 | p.Asp309Tyr | 1 | other | 1 | ||||||
| PH domain | 10 | p.Trp432Arg | 2 | PS* | 2 | |||||
| PH-REM union domain | 10 | p.Ser548Arg | 1 | PS | 1 | |||||
| 10 | p.Arg552Ser, p.Arg552Gly, p.Arg552Trp | 5 | PS* (4) | 5 | ||||||
| 10 | c.1330_1332del f | 1 | PS | 1 | ||||||
| RAF1 | CR2 domain | 7 | p.Ser257Leu | 5 | 9 | 0 | HCM* | 5 | ||
| 7 | p.Ser259Phe | 1 | HCM | 1 | ||||||
| 7 | p.Pro261His, p.Pro261Thr | 2 | HCM* | 2 | ||||||
| Activation domain (CR3) | 14 | p.Glu478Lys | 1 | HCM | 1 | |||||
| BRAF | CRD (CR1) | 6 | p.Gln257Arg | 2 | 5 | 0 | HCM, other | 2 | ||
| Activating domain (CR3) | 12 | p.Glu501Lys | 2 | PS* | 2 | |||||
| 14 | p.Asn581Asp | 1 | ||||||||
| 180 | 20 | 147 | 9 | |||||||
| Totals | 200 | 200 | 156 | |||||||
HCM, hypertrophic cardiomyopathy; PS, pulmonary valve stenosis.
a With some exceptions,e,f mutations detected in Spanish patients had been previously detected in other populations that included patients with Noonan or other NFCF syndromes. Intronic variants or polymorphisms described in coding regions are not included. Figure shows the types of syndromes presented by patients carrying mutations in different genes.
b Frequency of alterations in the different domains of the protein encoded by the PTPN11 gene. Due to the small number of carrier alleles, frequencies for other genes are not included.
c *denotes the type of heart disease present or predominant in patients with the alteration shown in the genotype column. When there was more than one type of associated heart disease, the number of patients with pulmonary valve stenosis (PS) is indicated in brackets.
d One patient who presented the p.Tyr63Cys mutation showed further impairment of exon 8, p.Met311Val in cys with the mutation, which cosegregated in relatives with the disease (3 cases). The P.Met311Val variant has not been described and, although not detected in 700 normal chromosomes (analyzed by partial sequencing of PTPN11), in silico studies 32,33 (see “Methods”) indicate that this is only a polymorphism.
e A new alteration was characterized in PTPN11, in familial form, that cosegregated with the phenotype and involved in-phase of the CAA triplet in exon 7. This glutamine (Gln) amino acid-encoding triplet is located adjacent to the Gln256 residue (CAA ins, Gln256) and was not detected in 700 normal chromosomes analyzed.
f One de novo patient presented the c.1330_1332del SOS1 mutation. This previously undescribed alteration was ruled out in 1000 normal chromosomes using high resolution melting. The manuscript describing it is currently in preparation.
The rate characterization in the PTPN11 study was 27% (172/643 patients). PTPN11's diagnostic yield increases to 43% (128/292) if only the patient group with heart disease is considered. Only 9 of 129 (7%) PTPN11 positive patients had no heart disease.
As regards the distribution of the mutations, 34% were in regulatory domains, 4% in bridge domains, and 62% in phosphatase domains. De novo and familial forms were detected in both the regulatory domain and the active site domain (Table 1). The most recurrent alteration, p.Asn308Asp (n=47), was located in the phosphatase domain, although the p.Asn308Ser change was also detected (n=9). In total, 107 mutant alleles were found in this domain. Mutations in the regulatory region of the active site domain were the second largest group (59 patients). The profile showed greater heterogeneity, with 12 residues involved, though they primarily affected exon 3.
KRASA KRAS gene study was initially proposed as the first choice in cases in which there were definite clinical grounds to suspect negative PTPN11, although no alterations were characterized in any case. It is now applied as a third- or fourth-level study in severe phenotypes associated with mental retardation.
SOS1Over the study period, 14 patients were characterized, all of whom had been diagnosed with NS-associated heart disease (primarily PS but also, occasionally, HCM). Exon 10 showed a high level of recurrence; alteration of arginine residue 552 was detected in 5 patients, with 3 types of amino acid changes p.Arg552Gly, p.Arg552Trp, and p.Arg552Ser. In this region, we detected a de novo alteration that had not been previously described: p.Asp430del (c.1330_1332del, formerly c.1330_1332delATG). This is an “in-phase” deletion involving aspartate 430, a highly conserved amino acid (manuscript in preparation). Affected relatives were detected in 3 of the 14 families with the SOS1 mutation.
RAF1In this series, 9 patients had mutations in RAF1; the most recurrent alteration (5 cases) was p.Ser257Leu. Notably, the mutation detected outside of this exon, the exon p.Asp478Lys 14, was the only one in this series that had been described in sporadic tumors as a de novo somatic mutation.9 It was confirmed that parents who were asymptomatic did not show this alteration, indicating that it was a de novo germline mutation. The p.Ser257Leu mutation was detected in the RAF1 gene in 2 unrelated patients from different hospitals and with suspected Costello syndrome (cases 6 and 9, Table 2).
Table 2. RAS-MAPK Syndrome Patients and Relatives for Whom Molecular Diagnosis Reoriented the Clinical Diagnosis.
| Case a | Family | Patientsindex cases | Initial diagnosis | Genotype | Final diagnosis |
| 1 | 1 | Index | Noonan | PTPN11-p.Thr468Met | LEOPARD |
| 2 | 2 | Index | Neurofibromatosis type I with HCM | PTPN11-p.Thr468Met | LEOPARD 16 |
| 3 | 3 | Index | Neurofibromatosis type I with HCM | PTPN11-p.Thr468Met | LEOPARD 16 |
| 4 | 4 | Index | Neurofibromatosis type I with HCM | PTPN11-p.Thr468Met | LEOPARD 16 |
| 5 | 5 | Index | Neurofibromatosis type I with HCM | PTPN11-p.Thr468Met | LEOPARD |
| 6 | 6 | Index | Costello c | RAF1-p.Ser257Leu | Noonan with HCM |
| 7 | 7 | Index | Noonan | BRAF-p.GlnQ257Arg | CFC |
| 8 | 8 | Index | Heart disease, no syndromic diagnosis | BRAF-p.GlnQ257Arg | LEOPARD |
| 9 | 9 | Index | Costello c | RAF1-p.Ser257Leu | Noonan with HCM |
| Family members | |||||
| 10 | 10 | Mother | Familial LEOPARD | PTPN11-normal | Unaffected, abundant freckles |
| 11 | 10 | Affected (index) | Familial LEOPARD | PTPN11-p.Asn308Asp | Noonan de novo |
| 12 | 11 | Mother (index) | Undiagnosed short stature and suggestive facies | PTPN11-p.Tyr63Cys | Noonan |
| 13 | 11 | Child b | Hidrops faetalis (neonatal death) | PTPN-p.Tyr63Cys | Noonan |
| 14 | 12 | Mother | Short stature and pterigium coli (suspected Turner) | PTPN-p.Asn308Asp | Noonan |
| 15 | 13 | Father | Cryptorchidism | PTPN-p.Pro491Thr | Noonan |
| 16 | 14 | Father | Bilateral cryptorchidism | SOS1-p.Arg552Trp | Noonan |
| 17 | 15 | Mother | Short stature and mildly suggestive phenotype | PTPN11-normal | Unaffected |
| 18 a 26 | Other family members d | No consultation, clinical status currently being assessed | 7 PTPN+ y 2 SOS1+ | Noonan |
CFC, cardio-facio-cutaneous syndrome; HCM, hypertrophic cardiomyopathy.
a Only cases in which the change in diagnosis has been documented are included here, so selection may not be exhaustive.
b Necropsy, kidney and liver tissues.
c Two unrelated patients from different hospitals with suspected Costello syndrome presented the p.Ser257Leu mutation in the RAF1 gene.
d Nine family members with a positive genotype in whom clinical signs were not reported.
The study of HRAS carried out in patients with suspected Costello syndrome ruled out recurrent alterations in all those analyzed during the study period. In 3 patients with this suspected diagnosis, mutations were characterized in some of the genes analyzed (Figure 1).
BRAFFive BRAF positive patients were detected. In 3 of those patients, the original clinical diagnosis of CFC was confirmed; in the other 2, the original diagnosis was modified based on the molecular findings (Table 2, cases 7 and 8).
The sum of the characterization rates obtained for each exon analyzed reached 63% in studies stratified by phenotype, a figure which is consistent with the range described in the literature. Increasing the number of genes analyzed improves characterization, albeit with decreasing efficiency: 20% (16/81), 13% (5/38) and 6% (1/15) with 2, 3, and 4 genes, respectively. However, in the group analyzed for 5 genes (6 cases), 2 patients were characterized, one of whom was positive for SOS1, which has now displaced KRAS and is the second choice in molecular analysis.
Genotype-Phenotype RelationshipsAll genes and all molecular alterations were associated with both PS and HCM in at least some patients (Table 1). Nevertheless, whereas PS appeared in all cases, HCM was usually (though not exclusively) limited to RAF1 and the specific PTPN11 mutation, p.Thr468Met. PS was the heart condition most frequently associated with the recurrent PTPN11 alteration, p.Asn308Asp (33 of 40 patients with reports of clinical signs); only 1 patient had HCM, and 3 had no heart disease. The RAF1 mutations were observed more frequently in patients with HCM (9/40 with HCM vs 0/28 PS) and in patients with LEOPARD (2/12). The p.Thr468Met mutation was very strongly associated with the LEOPARD phenotype and in some patients the molecular data even predicted the clinical diagnosis of LEOPARD, as the café au lait spots and lentigines typical of the condition only appeared later (Table 2, case 1). Interestingly, although the acronym points toward PS, in our series about half of the patients with this suspected diagnosis showed HCM. In 3 patients reported by Carcavilla et al.,19 the initial diagnosis was of neurofibromatosis type I.
Hematological abnormalities were primarily found to be associated with the PTPN11 exon 3; some were neonatal diagnoses.30 However, none of our patients with the typically associated Thr73 amino acid mutation25 had shown these processes, although all were pediatric patients.
The genotypic and phenotypic overlap of neuro-cardio- facio-cutaneous syndromes appears in Figure 1, which shows the number of patients suffering from different syndromes for each gene. In at least 9 index patients, the mutation identified led to a modification in the suspected diagnosis due to which patients had been referred (Table 2).
Family StudiesFamilial forms were detected in patients with mutations in PTPN11 (n=17, 11 in the coding region of the active site domain, and 6 in the regulatory region) and SOS1 (n=3) (Table 1). A total of 30 mutation-carrying family members were detected in those 20 families. Maternal inheritance was most frequent, with 14 pediatric index cases in which the mother was the carrier, and 1 adult patient whose daughter was affected. In one index patient, the diagnosis was suspected in the mother as a result of perinatal death. Molecular confirmation of the mutation in the mother preceded the post-mortem study which confirmed the diagnosis (Table 2, case 13). Other familial cases detected corresponded to 7 siblings in 6 families, and an uncle and grandfather, both maternal. In 1 family (mother and child carriers), a study of the grandparents showed that the mutation had occurred de novo in the previous generation.
As we have seen, cardiopathy is an almost constant finding in the index case mutation carriers, but was infrequently diagnosed and/or evaluated in parents with the alteration. Four patients had clinical signs that had not led to diagnosis (short stature, cryptorchidism) and in 9 cases there had been no consultation, diagnosis, or evaluation prior to the molecular findings (Table 2). Heart disease had only been diagnosed previously in 3 of the 19 positive adults detected in the family study.
Diagnostic Yield by Center and PhysicianThe diagnostic yield obtained was not homogeneous and there were differences between requesting hospitals. Table 3 presents data on centers requesting studies for at least 9 patients (54% maximum rate). Although the data are not shown by professional, some did exceed this percentage (9/14, 64.3%; P=.01).
Table 3. Diagnostic Yield of the Molecular Study Obtained for Patient Samples From Selected Hospitals Nationwide With at Least 9 Requests Over the 2005-2010 Study Period.
| Hospital a | Studies requested b | Only index patients | Positive cases c | Positive index patients | Yield d |
| 1 | 137 | 105 | 34 | 30 | 28.6% |
| 2 | 100 | 70 | 19 | 15 | 21.4% |
| 3 | 51 | 49 | 17 | 17 | 34.7% |
| 4 | 53 | 44 | 13 | 13 | 29.5% |
| 5 | 46 | 44 | 12 | 11 | 25% |
| 6 | 55 | 32 | 20 | 17 | 53.1% |
| 7 | 33 | 30 | 8 | 6 | 20% |
| 8 | 29 | 26 | 11 | 11 | 42.3% |
| 9 | 28 | 25 | 2 | 2 | 8% |
| 10 | 31 | 24 | 4 | 4 | 16.7% |
| 11 | 26 | 22 | 9 | 9 | 40.9% |
| 12 | 35 | 19 | 11 | 7 | 36.8% |
| 13 | 30 | 19 | 9 | 9 | 47.4% |
| 14 | 20 | 17 | 3 | 2 | 11.8% |
| 15 | 18 | 15 | 6 | 5 | 33.3% |
| 16 | 16 | 13 | 4 | 5 | 38.5% |
| 17 | 12 | 11 | 5 | 5 | 45.5% |
| 18 | 9 | 6 | 4 | 3 | 50% |
| Total | 571 | 171 | 34% |
a Ordered by the number of studies requested by the center in question over the 2005-2010 period.
b Includes all samples analyzed, index patients, and affected and unaffected family members.
c Refers to the total number of patients and family members who were mutation carriers.
d Yield is calculated from the number of index patients analyzed and characterized (positive) because studies of parental DNA, requested after genetic counseling but where a suggestive phenotype is absent, are often negative in both parents. The yield is slightly higher than that seen overall, which may reflect the higher levels of experience (due to a wider variety of cases) of these centers.
The overall yield of the molecular studies (200/643; 31%) increased by 50% (147/292) if only patients with heart disease were taken into account, and by 62% (134/218) if only PS and/or HCM were considered. PS and/or HCM were the most frequent types of heart disease in patients with mutation (139/156; 89%), while the spectrum of heart disease was more diverse and the percentage of disease represented by PS and/or HCM considerably lower in patients with no mutation (84/147 [54%]; P<.0001). The absence of heart disease, a rarity in the group of patients with mutation (9/156), was less uncommon in patients without mutation (101/343; P<.0001). There was a lower diagnostic yield in the group of patients for whom phenotypic data were not available (18 vs 31%; P=.0002). The exclusion of patients for whom information on heart disease was not available increased the diagnostic yield (156/400 [39%]; P=.01).
DiscussionThe molecular characterization of 230 Spanish patients with NS or other RAS-MAPK syndromes helped confirm the clinical diagnosis, and provided more accurate differential diagnoses. As Rodriguez-Marín observed,34 this more effective classification, supported by genomic assessment, can facilitate more targeted treatments.
The alterations detected in our population were mostly mutations which had already been described, a fact which facilitated the preparation of diagnostic reports. The distribution of mutations was similar to that reported in other populations.25, 26, 27, 28 Changes in certain amino acids (Asn308 in PTPN11, Arg552 in SOS1, and Ser257 in RAF1) were recurrent.
The observed genotypic profile does not differ from that described in other populations: PTPN11 is the gene most commonly affected, followed by SOS1. The association with HCM makes the study of RAF1 in PTPN11 negative patients imperative, and analysis should focus first on BRAF when there are signs of CFC. However, complementary analysis of these genes should not be ruled out in patients with other suspected RAS-MAPK syndromes; in 2 unrelated cases with suspected Costello, which is generally associated with HRAS,21 we detected the same mutation in RAF1. The contribution to differential diagnosis was particularly noticeable in neonates30 and in LEOPARD.19 The stratified study is consistent with the current recommendations from Romano et al.,29 who proposed a stratified approach based on the phenotype, in contrast to genotyping using arrays.
Molecular diagnosis helped to rule out the syndrome in some families who shared signs but who did not have the disease, and a familial form was confirmed post mortem. Of equal importance is the fact that family members with previously unrecognized heart disease were detected. It is noteworthy that, in 2 mutation-carrying parents, cryptorchidism was the only sign recorded in the medical history. As reported recently,35 survival in patients with NS and HCM is significantly reduced in comparison to nonsyndrome patients with HCM, and sometimes facial expressiveness does not help to detect affected individuals,19 making molecular analysis an essential diagnostic tool. Sanchez et al.3 recently reported the association between NS and HCM.
In patients with PS or HCM type heart disease, the diagnostic yield increased greatly, from 31% for the study population as a whole to 62%. The rate of characterization varied greatly across centers, even after excluding those requesting only a small number of studies. The overall diagnostic yield, which was similar to that described in series lacking rigorous selection,26 reached 64% in specific professionals, indicating that the molecular approach proposed was appropriate and that the establishment of preliminary diagnoses needs improving in Spain.
Factors contributing to the higher rate of genotypic characterization include patient assessment by expert dysmorphologists and the existence of heart disease. The Van der Burgt et al. clinical criteria,5 which are widely accepted and used, emphasize characteristic facies as a key diagnostic tool. Our data from patients with a positive genotype highlight the relevance of the presence and type of heart disease; because this is a more objective and uniform criteria it may have led, in this context, to a more precise preliminary diagnosis.
Heart disease is sometimes the reason for a first visit in patients with NS, and it is essential that cardiologists suspect, and are sensitized to, an inherited syndrome that often appears de novo. The fact that it appears de novo means that a family phenotype will not be available to support the suspected diagnosis. It is therefore essential to run appropriate complementary tests; molecular studies have shown that they can provide diagnostic confirmation, and hence support to family members. We are working on optimizing other markers that may help to provide a more objective basis for clinical suspicion, such as photos of facial morphometry36 and the detection of overexpression of RAS-MAPK pathway messengers.37
Two important limitations of the study are that patients came from different centers and that only a limited amount of clinical data was considered. The study's main strength is the number of patients included. The comprehensive collection of clinical data which would allow for an investigation of genotype/phenotype correlations is beyond the scope of this study. Moreover, the selection of patients with detailed data could have biased the results, though we believe that our approach also allowed us to obtain a more accurate picture of the situation in Spain.
ConclusionsThere is a certain amount of variability in the way in which suspected diagnoses of neuro-cardio- facio-cutaneous syndromes are handled in Spain; molecular data were key to better diagnoses and to the detection of familial cases. The high degree of association with documented heart disease, coupled with the discovery of cases in which no cardiac study had been performed, should alert us to the importance of carrying out the appropriate studies and examinations in patients and family members.
FundingFunded by Fondo de Investigaciones Sanitarias (grant number PI 06/1179).
Conflict of interestNone declared.
Acknowledgements
The authors would like to thank the patients and relatives who participated as well as the different departments and units that shared their samples with us. We would particularly like to mention doctors Zapico (Hospital de Alicante), Lapunzina (Hospital La Paz), Lopez-Siguero (Hospital Carlos Haya), Del Campo (Hospital Vall d’Hebron), Tamarit and Pozo (Hospital Niño Jesús), Rodrigo-Palacios (Hospital General Yagüe), Galán (Hospital Perpetuo Socorro), Guitart (Hospital Parc Taulí), Martínez (Hospital Reina Sofía), García-Cuartero and Carrasco (Hospital Severo Ochoa), Luzuriaga (Hospital Universitario Marqués de Valdecilla), Lautre (Hospital Clínico San Carlos), Herrera (Complejo Hospitalario de Jaén), and Albiñana (Hospital de Almansa). Their cases were especially relevant to this study, either because of their number or because of their salient features. Begoña Ezquieta is a researcher of the U753 of CIBERER (Biomedical Network Research on Rare Diseases).
SUPPLEMENTARY MATERIAL.Supplementary material associated with this article can be found, in the online version, available at doi:10.1016/j.rec.2011.12.017.
Appendix A. Supplementary dataReceived 11 July 2011
Accepted 14 December 2011
Corresponding author: Laboratorio de Diagnóstico Molecular, Hospital Infantil, Hospital General Universitario Gregorio Marañón, Dr. Esquerdo 46, 28007 Madrid, Spain. bezquieta.hgugm@salud.madrid.org