Cardiac amyloidosis is a disease caused by the extracellular deposition of abnormal insoluble fibrils in the heart. The 2 main types are primary amyloidosis, due to light-chain deposits, and transthyretin amyloidosis (ATTR), with its 2 forms: hereditary (hATTR) (due to mutations in the TTR gene) and wild type (ATTRwt). In hATTR, more than 120 mutations have been described, such as Val50Met (the most common, associated with familial amyloid polyneuropathy) and Val142Ile (predominantly cardiac phenotype).1 It is important to identify patients whose amyloidosis is due to a genetic defect, as it changes the treatment and is also highly relevant for their relatives.2 ATTRwt affects almost exclusively the heart and is severely underdiagnosed. Its frequent association with aortic stenosis (AS) has been described, with a prevalence of ATTRwt of between 5.3% and 16% in patients with AS, in whom it confers a poor prognosis.3 There are now treatments for ATTR that are changing its prognosis.
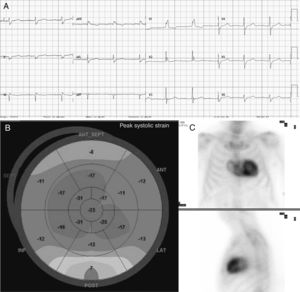
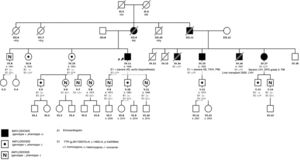
We present a large family of Mallorcan origin. The proband was a 72-year-old man under outpatient follow-up for AS. He had a long history of hypertension, dyslipidemia, type 2 diabetes mellitus, as well as a 10-year history of chronic renal failure due to interstitial nephropathy of unknown etiology. He had undergone bilateral hand tendon surgery (trigger finger) at the age of 60 years. He was on treatment with torasemide 5mg/d, metformin, sitagliptin and atorvastatin. At follow-up he was found to have symptomatic AS with dyspnea on moderate exertion. On examination, blood pressure was 110/60mmHg, heart sounds were regular, there was a IV/VI systolic murmur in the aortic area, normal vesicular breath sounds, and no signs of heart failure. Electrocardiography (figure 1) showed sinus rhythm, long PR interval, right bundle branch block, inferior Q waves, and low voltage in the limb leads. Echocardiography showed moderate concentric left ventricular hypertrophy (15mm) with preserved ejection fraction and features of severe AS (area, 0.8cm2; maximum/mean gradient, 67/42mmHg). The aortic valve was replaced with a bioprosthetic valve, without complications. At follow-up, the possibility of concurrent ATTR and AS was considered, for several reasons: a) male patient older than 65 years; b) electrocardiogram showing pseudoinfarction pattern (inferior Q waves in the absence of coronary artery disease), conduction defect (right bundle branch block) and low voltages (discordance with ventricular hypertrophy); c) history of stenosing tenosynovitis of the hand (associated with carpal tunnel); and d) “cured” chronic hypertension (blood pressure 110/60 mmHg on only 5 mg torasemide). Echocardiography was repeated at this point with longitudinal strain (figure 1), which showed the typical amyloid pattern with loss of strain in the basal segments and apical preservation. 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) myocardial scintigraphy (figure 1) showed grade 3 cardiac uptake and confirmed the diagnosis of ATTR, after it was determined that light chains and blood and urine immunofixation were negative. Gene sequencing for TTR found the Val50Met mutation in heterozygosis. Assessment by neurology and internal medicine found a small fiber neuropathy with autonomic dysfunction (Sudoscan with pathological findings and erectile dysfunction), and nephrology concluded that the patient’ chronic renal disease was glomerular and interstitial disease due to transthyretin deposits. Finally, a family study was performed (figure 2), showing that 1 male cousin also had AS treated with transcatheter aortic valve implant and pacemaker for complete atrioventricular block, who after evaluation was also found to have hATTR (moderate hypertrophy with grade 3 DPD), 1 female cousin with hATTR and pacemaker (in both cousins, primary amyloidosis was ruled out), and another 2 cousins and 1 uncle of the proband who had died due to familial amyloid polyneuropathy. The proband's mother, who died from heart disease, was considered to be an obligate carrier. Thirteen other relatives were assessed clinically and genetically: 6 were asymptomatic carriers (being followed up) and 6 were not carriers.


Several studies have described the association between AS and ATTR, but almost always in relation to the wild type 1,3(1 single case with Val142Ile4). This is the first family with the Val50Met mutation in which this concomitance is described. It is a large family with 2 members with severe AS and hATTR, and with several affected members with a predominantly cardiac, neurological or mixed phenotype. Although the pathophysiology of the association between AS and ATTR is unclear, it has been postulated that amyloid deposition in the aortic valve could trigger or contribute to the development of AS, although we cannot rule out that these 2 diseases were independent and their coexistence could have been due to their high prevalence in those older than 65 years. The coexistence of 2 types of amyloid (hereditary and wild type) in the heart is also possible. It is important to highlight that the presence of AS should alert the clinician to look for signs suggestive of ATTR (eg, clinical features, history, electrocardiography, echocardiography),5 for several reasons: a) it changes the treatment, as certain drugs should be avoided (calcium antagonists, digoxin, beta blockers, angiotensin-converting enzyme inhibitors); b) ATTR can act as a modifier of AS and produce a more severe phenotype (more heart failure and arrhythmias), and the deposits may affect other organs, with some authors even preferring transcatheter aortic valve implantation for these patients, given their increased surgical risk1; c) patients may benefit from existing approved ATTR-specific drugs that change the course of the disease; d) diagnosis with cardiac scintigraphy is simple; and e) if it is hATTR, 50% of the patient's relatives may be at risk, and they should be identified early.
FUNDINGInstituto de Investigación Sanitaria de Palma (IdISBa), Palma de Mallorca, Balearic Islands, Spain.