Hereditary transthyretin amyloidosis is an autosomal dominant disease caused by mutations in the transthyretin gene. Of the more than 100 such mutations reported, Val30Met is the most common; the condition is called familial amyloidotic polyneuropathy (FAP) or Corino de Andrade disease in patients with predominant neurological damage.1 Identification of patients whose amyloidosis is due to a genetic defect is vital because such information affects the treatment strategy and is of great importance for relatives.2 Although its worldwide prevalence is low, various endemic foci have been described. The island of Mallorca currently has the fifth highest number of affected individuals, behind Portugal, Sweden, Japan, and Brazil. In addition, Spain has another endemic focus, albeit smaller, in Valverde del Camino (Huelva).3 Individuals with the Val30Met mutation generally present with peripheral neuropathy and progress to autonomic and motor neuropathy, with late onset of cardiac conduction disorders and without cardiac hypertrophy.4 Currently approved treatments for FAP include liver transplant and tafamidis, a drug that stabilizes transthyretin. The other drugs under study show promising initial results. Current recommendations are to begin drug therapy or consider liver transplant at the first appearance of neurological signs and symptoms. Cardiac amyloidosis is one of the main causes of death in FAP but many of these patients with cardiac involvement are underdiagnosed.5
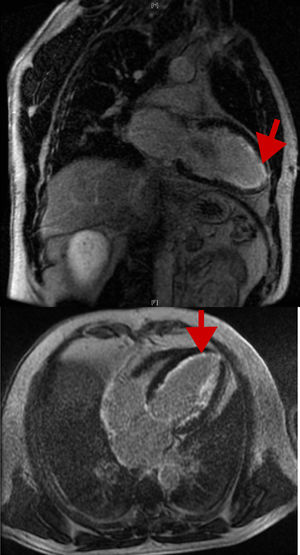
Our objective was to evaluate cardiac involvement in a large series of patients with FAP because this aspect of the disease is poorly characterized in the literature due to its low prevalence, particularly in Spain, for which there are no published data. We reviewed the medical records of patients with FAP (positive genetic study findings for the Val30Met mutation in the transthyretin gene in all patients and the presence of amyloid in subcutaneous fat, rectal, and salivary gland biopsy in all symptomatic patients). Demographic, clinical, electrocardiographic, echocardiographic, and Holter monitoring data were collected, as well as cardiac magnetic resonance imaging and diphosphonate scintigraphy data if they had been performed. Cardiac involvement was defined as the presence of specific signs or symptoms, arrhythmias, atrioventricular (AV) conduction disorders, left ventricular hypertrophy on electrocardiography (ECG) or echocardiography, or late enhancement on cardiac magnetic resonance imaging. Data from 132 patients were analyzed (Table): 104 symptomatic carriers (78.8%) and 28 asymptomatic carriers (21.2%). The mean ages were 47.4±17 years at diagnosis and 57.2±16.4 years at follow-up; 69 (52.2%) were men. Of the symptomatic carriers, 83 (79.8%) had polyneuropathy and 56 (53.8%) had some symptom of cardiovascular or cardiac involvement: 15% had palpitations; 10.4%, dyspnea; 4%, syncope; 20.6%, dysautonomia symptoms; and 9.8%, heart failure. The ECG was pathological in 39 patients (36%), due to signs of left ventricular hypertrophy, arrhythmias, or AV conduction changes: 13 (9.8%) with sinus node dysfunction or atrial fibrillation (bradycardia-tachycardia syndrome), 17 (12.9%) with different degrees of AV block, and 12 (9.1%) with His bundle-branch block. The mean maximum thickness of the left ventricular wall was 11.2±3.2mm, with a left ventricular ejection fraction of 61±6%; 19 patients (23.5%) had a left ventricular wall thickness ≥ 11mm and 10 (12.3%) had a thickness ≥ 15mm. Of these 10, 5 underwent cardiac magnetic resonance imaging, with 3 showing late gadolinium enhancement in the subendocardial region in the characteristic ring shape (Figure). In addition, 46% had diastolic dysfunction and 9 (6.8%) required pacemaker implantation during follow-up due to sinus node dysfunction or advanced AV block. Liver transplant was performed in 54 patients (41%), as well as 1 combined heart-liver transplant; 11 patients received tafamidis in recent years. During follow-up, 22 patients (16.7%) died.
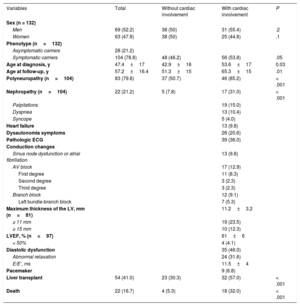
Clinical Data of the Patients Included in the Study
| Variables | Total | Without cardiac involvement | With cardiac involvement | P |
|---|---|---|---|---|
| Sex (n = 132) | ||||
| Men | 69 (52.2) | 38 (50) | 31 (55.4) | .2 |
| Women | 63 (47.8) | 38 (50) | 25 (44.6) | .1 |
| Phenotype (n=132) | ||||
| Asymptomatic carriers | 28 (21.2) | |||
| Symptomatic carriers | 104 (78.8) | 48 (46.2) | 56 (53.8) | .05 |
| Age at diagnosis, y | 47.4±17 | 42.9±16 | 53.6±17 | 0.03 |
| Age at follow-up, y | 57.2±16.4 | 51.3±15 | 65.3±15 | .01 |
| Polyneuropathy (n=104) | 83 (79.8) | 37 (50.7) | 46 (85.2) | < .001 |
| Nephropathy (n=104) | 22 (21.2) | 5 (7.8) | 17 (31.0) | < .001 |
| Palpitations | 19 (15.0) | |||
| Dyspnea | 13 (10.4) | |||
| Syncope | 5 (4.0) | |||
| Heart failure | 13 (9.8) | |||
| Dysautonomia symptoms | 26 (20.6) | |||
| Pathologic ECG | 39 (36.0) | |||
| Conduction changes | ||||
| Sinus node dysfunction or atrial fibrillation | 13 (9.8) | |||
| AV block | 17 (12.9) | |||
| First degree | 11 (8.3) | |||
| Second degree | 3 (2.3) | |||
| Third degree | 3 (2.3) | |||
| Branch block | 12 (9.1) | |||
| Left bundle-branch block | 7 (5.3) | |||
| Maximum thickness of the LV, mm (n=81) | 11.2±3.2 | |||
| ≥ 11 mm | 19 (23.5) | |||
| ≥ 15 mm | 10 (12.3) | |||
| LVEF, % (n=97) | 61±6 | |||
| < 50% | 4 (4.1) | |||
| Diastolic dysfunction | 35 (46.0) | |||
| Abnormal relaxation | 24 (31.6) | |||
| E/E’, ms | 11.5±4 | |||
| Pacemaker | 9 (6.8) | |||
| Liver transplant | 54 (41.0) | 23 (30.3) | 32 (57.0) | < .001 |
| Death | 22 (16.7) | 4 (5.3) | 18 (32.0) | < .001 |
AV, atrioventricular; ECG, electrocardiography; LV, left ventricle; LVEF, left ventricular ejection fraction.
.")
Although the cardiac involvement was clinically evident only in the late phase of the disease (these patients were 10 years older than those who only had the neurological phenotype), it was associated with greater comorbidity and was significantly linked to increased mortality (P = .003). On multivariable analysis, the parameters independently related to mortality during follow-up (P < .05) were pathological ECG, AV block, heart failure, pacemaker, neurological involvement, and renal failure. Mean duration from symptom onset to death was 7.3 years.
In conclusion, our results, based on a relatively large series given the low prevalence of the disease, provide additional information on FAP and help to describe its demographic characteristics, clinical presentation, diagnosis, and clinical course. FAP is a rare disease that not only causes classic neurological symptoms, as has been thought for many years, but also has a relatively frequent cardiovascular involvement, particularly pathological ECG, rhythm disturbances and AV conduction disorders, left ventricular hypertrophy with diastolic dysfunction, and dysautonomia symptoms, and is associated with increased morbidity and mortality. Compared with Portugal, Sweden, and Japan, the endemic area of Mallorca shows more frequent left ventricular hypertrophy and heart failure and worse prognosis. Cardiologists should perform close follow-up of these patients–with annual ECG, echocardiography, and Holter monitoring–and expand the evaluation (eg, cardiac magnetic resonance imaging, diphosphonate scintigraphy) in the presence of symptoms or changes.
FUNDINGCIBEROBN (CB12/03/30038), Madrid, Spain.