Many neuromuscular diseases affect the heart and, although involvement is usually asymptomatic, it can occasionally be clinically relevant. Examples of such neuromuscular diseases include muscular dystrophy; congenital, inflammatory, and metabolic myopathies; certain hereditary degenerative diseases; diseases of the neuromuscular junction, and acquired or hereditary peripheral neuropathies.1 Correct diagnosis requires a high degree of clinical suspicion and an experienced multidisciplinary team who coordinate skeletal muscle and peripheral nervous system study including determination of muscle enzymes, neurophysiological study, and, if necessary, muscle biopsy. An exhaustive assessment of respiratory function is recommended, particularly if patients present with clinical abnormalities, as this may help establish prognosis or be the main clinical manifestation. The most prevalent heart abnormalities are cardiomyopathies, ventricular arrhythmias, and conduction system disorders associated with sudden cardiac death.2 However, the evidence available is limited to series of isolated patients, and clinical guidelines are based on expert recommendations.3
This article describes cardiac manifestations and familial penetrance in our series of patients with a genetic diagnosis.
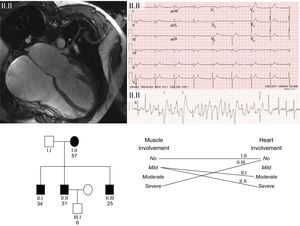
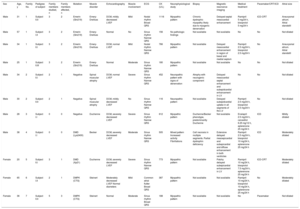
The study included 11 patients, with a mean age of 36.18 ± 13.4 years and a predominance of men (72%), from 7 families. The The clinical and genetic findings for the 11 patients are summarized in the Table. The most frequent clinical expression was dilated cardiomyopathy with left ventricular systolic dysfunction. There was a high prevalence of rhythm disorders (81.8%). The prevalence of rhythm disorders was highest in patients with Steinert disease and Emery-Dreifuss muscular dystrophy: all patients with Steinert disease had rhythm disorders and 75% had Emery-Dreifuss muscular dystrophy.2 One patient had a family history of sudden death. The Figure shows an example of a family with Emery-Dreifuss muscular dystrophy.
Clinical and Genetic Characteristics of the 11 Patients
| Sex | Age, y | Family No. | Pedigree of subject | Family members studied, n | Family members affected, n | Mutation | Muscle disorder | Echocardiography | Muscle involvement | ECG | CK (U/L) | Neurophysiological study | Biopsy | Magnetic resonance imaging | Medical treatment | Pacemaker/CRT/ICD | Atrial size |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male | 31 | 1 | Subject II.II | 3 | 2 | Emerin (Gln219) | Emery-Dreifuss | DCM, mildly decreased LVEF | Mild | Nodal rhythm Broad QRS | 1116 | Myopathic pattern | Chronic dystrophic myopathy likely due to myogenic denervation | Delayed septal mesocardial enhancement | Ramipril 5 mg/24 h, bisoprolol 5 mg/24 h | ICD-CRT | Aneurysmal atrium. Atrial standstill |
| Male | 57 | 1 | Subject I.II | 3 | 2 | Emerin (Gln219) | Emery-Dreifuss | Normal | No | Sinus rhythm Narrow QRS | 156 | No pathologic findings | Not available | Not available | No | No | Not dilated |
| Male | 34 | 1 | Subject II.I | 3 | 2 | Emerin (Gln219) | Emery-Dreifuss | DCM, normal LVEF | Mild | Nodal rhythm Narrow QRS | 766 | Myopathic pattern | Not available | Delayed mesocardial enhancement in region of basal and medial septum | Ramipril 2.5 mg/24 h, bisoprolol 2.5 mg/24 h | No | Aneurysmal atrium. Atrial standstill |
| Male | 25 | 1 | Subject II.III | 3 | 2 | Emerin (Gln219) | Emery-Dreifuss | Normal | Moderate | Sinus rhythm Narrow QRS | 180 | Myopathic pattern | Not available | Not available | No | No | Not dilated |
| Male | 34 | 2 | Subject II.I | 2 | 1 | Negative | Spinal muscular atrophy | DCM, normal LVEF | Severe | Sinus rhythm Narrow QRS | 452 | Neuropathic pattern with signs of denervation | Atrophy with neurogenic component | Delayed mesocardial septal enhancement and subepidcardial enhancement in LV | No | No | Not dilated |
| Male | 30 | 2 | Subject II.II | 2 | 1 | Negative | Spinal muscular atrophy | DCM, mildly decreased LVEF | No | Sinus rhythm Narrow QRS | 116 | Neuropathic pattern | Not available | Delayed subepidcardial uptake in all segments of the LV | Ramipril 2.5 mg/24 h, bisoprolol 2.5 mg/24 h | No | Not dilated |
| Male | 28 | 3 | Subject I.I | 0 | 0 | Negative | Duchenne | DCM, severely decreased LVEF | Severe | Sinus rhythm Narrow QRS | 612 | Myopathic pattern | Duchenne/Becker phenotype, predominantly Duchenne | Not available | Enalapril 2.5 mg/24 h, carvedilol 6.25 mg/12 h, eplerenone 25 mg/24 h | ICD | Mildly dilated |
| Male | 38 | 4 | Subject I.I | 0 | 0 | DMD (Lys2400) | Becker | DCM, severely decreased LVEF | Moderate | Sinus rhythm Narrow QRS | 505 | Mixed pattern Increased activity Fibrillations | Cell necrosis in multiple segments. Partial dystrophin deficiency | Extensive delayed mesoepicardial and subepicardial and diffuse enhancement in both ventricles | Ramipril 2.5 mg/24 h, bisoprolol 10 mg/24 h, eplerenone 25 mg/24 h | ICD | Moderately dilated |
| Female | 20 | 5 | Subject II.I | 3 | 1 | DMD (Xp21) | Duchenne | DCM, severely decreased LVEF | Severe | Sinus rhythm Narrow QRS | 773 | Myopathic pattern | Not available | Patchy delayed subepicardial enhancement in LV | Ramipril 10 mg/24 h, bisoprolol 7.5 mg/24 h, eplerenone 25 mg/24 h | ICD-CRT | Moderately dilated |
| Female | 65 | 6 | Subject I.II | 2 | 1 | DMPK (CTG) | Steinert | Moderately decreased LVEF Normal diameters | Mild | Common atrial flutter Broad QRS | 387 | Myopathic pattern | Not available | Not available | Ramipril 10 mg/24 h, bisoprolol 10 mg/24 h, eplerenone 25 mg/24 h | No | Moderately dilated |
| Female | 36 | 7 | Subject II.II | 5 | 3 | DMPK (CTG) | Steinert | Normal | Moderate | Sinus rhythm Broad QRS | 188 | Myopathic pattern | Not available | Not available | No | Pacemaker | Not dilated |
CK, creatine kinase; CRT, cardiac resynchronization therapy; DCM, dilated cardiomyopathy; ECG, electrocardiogram; ICD, implantable cardioverter-defibrillator; LV, left ventricle; LVEF, left ventricular ejection fraction.

Family with Emery-Dreifuss muscular dystrophy with a novel mutation in the gene encoding emerin. The upper left image shows the cardiac magnetic resonance image of the index case with severe dilatation of both atria. The upper right image shows the resting electrocardiogram with nodal rhythm due to atrial standstill and development of polymorphic tachycardia in the exercise electrocardiogram. The lower part of the figure shows the family tree. Note the limited correlation between the degree and severity of heart involvement and skeletal muscle involvement in the 4 carriers of the mutation. Squares correspond to men and circles to women. Shading shows affected individuals and no shading represents healthy individuals.
Most of the patients (81.8%) had some degree of muscle damage, with mild, moderate, or severe involvement reported in 27.3% of patients each. In our sample, there was no statistically significant relationship between muscle damage recorded as mild, moderate, or severe (according to symptoms, clinical signs, and neuromuscular examination with electromyographic study performed by the neurology department) and cardiac involvement recorded as a mild, moderate, or severe decrease according to ejection fraction (Spearman correlation, ρ = 0.301; P > .05). Likewise, there was no significant relationship between creatine kinase (CK) and cardiac involvement (ρ = 0.54; P > .05) or between CK and muscle damage (ρ = 0.43; P > .05).
In the case of respiratory involvement, 18% of patients needed specialist respiratory follow-up (1 patient required invasive mechanical ventilation).
Overall, 54.5% of the patients were receiving medical treatment for heart failure with β-blockers and angiotensin converting enzyme inhibitors, and aldosterone antagonists or diuretics added as necessary. Thirty-six percent had an implantable cardioverter-defibrillator and of these, 50% had resynchronization. Nine per cent had a pacemaker without defibrillation or resynchronization.
With regards familial relationships, the index cases led to study of 15 additional family members. Of these, 8 were affected; 3 probands did not show a causal mutation in the genetic test.
There are limited recommendations with a low level of evidence for the treatment of these patients.3 For example, stratification of arrhythmic risk may be performed for an implantable cardioverter-defibrillator or pacemaker. In patients with Steinert disease, limb-girdle type 1B dystrophy, or Emery-Dreifuss muscular dystrophy, an implantable cardioverter-defibrillator is recommended when ventricular arrhythmias are present or pacing is indicated, with a level of evidence and grade of recommendation II B.4 In addition, in limb-girdle dystrophy, Steinert disease, and Kearns-Sayre syndrome, implantation of a permanent pacemaker is considered in the event of any atrioventricular block, even first-degree block. Our data support this approach, as they show a high prevalence of rhythm disorders. Another aspect under debate is whether anticoagulation is needed in patients with atrial standstill (typical in Emery-Dreifuss muscular dystrophy) or when supraventricular arrhythmias are present. In 1 of our families, such arrhythmias occurred in 2 affected men, with increased atrial volumes and no atrial activity in the electrocardiogram (ECG) or intracardiac ECG. This situation could predispose the patient to a higher risk of stroke, but there is no evidence to support a recommendation for oral anticoagulation in these patients.
No statistically significant relationships were observed in our series, probably due to the small sample size, although a weak correlation was observed between the severity of muscle damage and cardiac involvement. However, our results may be biased because, in disorders such as Emery-Dreifuss muscular dystrophy, cardiac involvement may precede muscle damage. The availability of genetic testing allows cascade screening of family members to identify individuals with potentially severe cardiac involvement who may benefit from early treatment even though no skeletal muscle damage is evident.
Patients with the most severe cardiac manifestations are referred more frequently to our department, constituting a selection bias. For this reason, caution should be exercised when drawing general conclusions on the prevalence or type of cardiac involvement.
Patients diagnosed with neuromuscular damage should undergo cardiovascular screening through assessment of medical and family history and the performance of physical examination, ECG, and echocardiography. For patients without cardiac involvement, regular visits with ECG and echocardiography are recommended at intervals adapted to the characteristics of each individual patient.3 The cardiologist should reassess the patient when new symptoms or disorders emerge in the tests undertaken. First-degree family members should be screened at the time of diagnosis.3
In conclusion, because of their complexity and low prevalence, neuromuscular diseases require closer clinical follow-up and management by experienced multidisciplinary teams, which should include a cardiologist.